| |
Programs are released by the Pollock Laboratory under a "use at your own risk" policy. They are mostly designed as research instruments rather than as user-friendly processed applications. They are generally written in C (some with perl wrappers and text-processors, and some with complementary R script for visualization of results) and operate using control files. Usually the graduate students(s) or postdoc(s) who have written the programs are likely to be most helpful in getting them to work. |
|
|
|
SchistoGenomics: A page for Schistosome genomics-related scripts,
programs, and data.
Ancestral TE: A program to infer relationships among replicating ancestral TE sequences in
a TE family.
SSRClouds: A program to empirically find clouds of sequence to
detect short sequence repeats.
Phylogenetics, Likelihood, Evolution, and CompleXity: A program for rapid likelihood analysis on large phylogenies using partial sampling of substitution histories.
Identify repeat structure in large eukaryotic genomes using oligonucleotide counts. It works efficiently on a single desktop computer with 1 Gb memory.
Use high throughput sequencing to simultaneously analyze binding dissociation constants for large repertoires of sequences.
Three programs used to analyze coevolution of transcription factor binding sites.
Detect pairwise coevolutionary among residues in a set of proteins
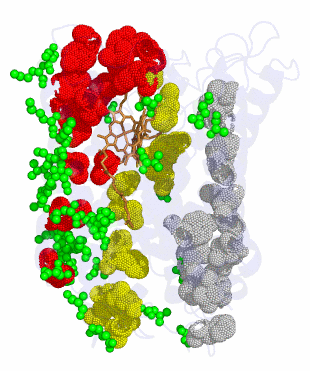
using likelihood ratio tests. Note: This link is largely historical. Updates to this program have since been made and are available upon request from Zhengyuan Wang. Myoglobin data used in the 1999 paper are also available.
Convergence/Divergence code from Castoe et al 2009 for use with codeml.
Code and really minimal explanation from from Pollock and Larkin 2004 and Hentges et al 2007. Apologies for the lousy web page transfer.
|
|