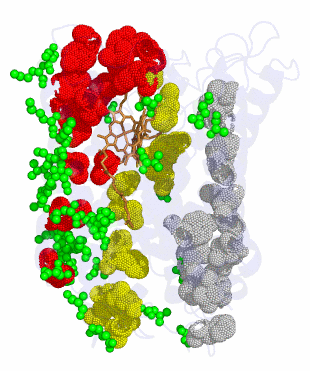
SP transcription factor paralogs and DNA binding sites coevolve and adaptively converge in mammals and birds
Ken Daigoro Yokoyama and D. D. Pollock
Functional modification of regulatory proteins can affect hundreds of genes throughout the genome, and is therefore thought to be almost universally deleterious. This belief, however, has recently been challenged. A potential example comes from transcription factor SP1, for which statistical evidence indicates that motif preferences were altered in eutherian mammals. Here, we set out to discover possible structural and theoretical explanations, evaluate the role of selection in SP1 evolution, and discover effects on co-regulatory proteins. We show that SP1 motif preferences were convergently altered in birds as well as mammals, inducing coevolutionary changes in over 800 regulatory regions. Structural and phylogenic evidence implicates a single causative amino acid replacement at the same SP1 position along both lineages. Furthermore, paralogs SP3 and SP4, which co-regulate SP1 target genes through competitive binding to the same sites, have accumulated convergent replacements at the homologous position multiple times during eutherian and bird evolution, presumably to preserve competitive binding. To determine plausibility, we developed and implemented a simple model of transcription factor and binding site coevolution. This model predicts that, in contrast to prevailing beliefs, even small selective benefits per locus can drive concurrent fixation of transcription factor and binding site mutants under a broad range of conditions. Novel binding sites tend to arise de novo, rather than by mutation from ancestral sites, a prediction substantiated by SP1 binding site alignments. Thus, multiple lines of evidence indicate that selection has driven convergent evolution of transcription factors along with their binding sites and co-regulatory proteins.
Phylogenetics, Likelihood, Evolution and Complexity (PLEX)
A. P. Jason de Koning, Wanjun Gu, Todd A. Castoe, and D. D. Pollock
Summary: PLEX is a flexible and fast Bayesian MCMC software
program for large-scale analysis of nucleotide and amino acid data
using complex evolutionary models in a phylogenetic framework.
The program gains large speed improvements over standard approaches
by implementing 'partial sampling of substitution histories',
a data augmentation approach that can reduce data analysis times
from months to minutes on large comparative datasets. A variety of
nucleotide and amino-acid substitution models are currently implemented, including non-reversible and site-heterogeneous mixture
models. Due to efficient algorithms that scale well with data size and
model complexity, PLEX can be used to make inferences from hundreds
to thousands of taxa in only minutes on a desktop computer. It
also performs probabilistic ancestral sequence reconstruction. Future
versions will support detection of co-evolutionary interactions
between sites, probabilistic tests of convergent evolution, and rigorous
testing of evolutionary hypotheses in a Bayesian framework.
Availability and Implementation: PLEX v1.0 is licensed under
GPL€. Source code and documentation will be available for download
at www.evolutionarygenomics.com/ProgramsData/PLEX. PLEX is
implemented in C++ and supported on Linux, Mac OS X, and other
platforms supporting standard C++ compilers. Example data, control
files, documentation and accessory Perl scripts are available from
the website.
*Contact: David.Pollock@UCDenver.edu
Supplementary Information: Supplemental results file
Copyleft 2012. All rites reversed.
Amino acid coevolution induces an evolutionary Stokes shift
D. D. Pollock, G. Thiltgen, and R. A. Goldstein
The process of amino acid replacement in proteins is context-dependent, with substitution rates influenced by local structure, functional role, and amino acids at other locations. Predicting how these differences affect replacement processes is difficult. To make such inference easier, it is often assumed that the acceptabilities of different amino acids at a position are constant. However, evolutionary interactions among residue positions will tend to invalidate this assumption. Here, we use simulations of purple acid phosphatase evolution to show that amino acid propensities at a position undergo predictable change after an amino acid replacement at that position. After a replacement, the new amino acid and similar amino acids tend to become gradually more acceptable over time at that position. In other words, proteins tend to equilibrate to the presence of an amino acid at a position through replacements at other positions. Such a shift is reminiscent of the spectroscopy effect known as the Stokes shift, where molecules receiving a quantum of energy and moving to a higher electronic state will adjust to the new state and emit a smaller quantum of energy whenever they shift back down to the original ground state. Predictions of changes in stability in real proteins show that mutation reversals become less favorable over time, and thus, broadly support our results. The observation of an evolutionary Stokes shift has profound implications for the study of protein evolution and the modeling of evolutionary processes.
Transcriptome sequencing of black grouse (Tetrao tetrix) for immune gene discovery and microsatellite development
Wang B, Ekblom R, Castoe TA, Jones EP, Kozma R, Bongcam-Rudloff E, Pollock DD, Höglund J
The black grouse (Tetrao tetrix) is a galliform bird species that is important for both ecological studies and conservation genetics. Here, we report the sequencing of the spleen transcriptome of black grouse using 454 GS FLX Titanium sequencing. We performed a large-scale gene discovery analysis with a focus on genes that might be related to fitness in this species and also identified a large set of microsatellites. In total, we obtained 182 179 quality-filtered sequencing reads that we assembled into 9035 contigs. Using these contigs and 15 794 length-filtered (greater than 200 bp) singletons, we identified 7762 transcripts that appear to be homologues of chicken genes. A specific BLAST search with an emphasis on immune genes found 308 homologous chicken genes that have immune function, including ten major histocompatibility complex-related genes located on chicken chromosome 16. We also identified 1300 expressed sequence tag microsatellites and were able to design suitable flanking primers for 526 of these. A preliminary test of the polymorphism of the microsatellites found 10 polymorphic microsatellites of the 102 tested. Genomic resources generated in this study should greatly benefit future ecological, evolutionary and conservation genetic studies on this species.
The interface of protein structure, protein biophysics, and molecular evolution
Liberles DA, Teichmann SA, Bahar I, Bastolla U, Bloom J, Bornberg-Bauer E, Colwell LJ, de Koning AP, Dokholyan NV, Echave J, Elofsson A, Gerloff DL, Goldstein RA, Grahnen JA, Holder MT, Lakner C, Lartillot N, Lovell SC, Naylor G, Perica T, Pollock DD, Pupko T, Regan L, Roger A, Rubinstein N, Shakhnovich E, Sjölander K, Sunyaev S, Teufel AI, Thorne JL, Thornton JW, Weinreich DM, Whelan S
The interface of protein structural biology, protein biophysics, molecular evolution, and molecular population genetics forms the foundations for a mechanistic understanding of many aspects of protein biochemistry. Current efforts in interdisciplinary protein modeling are in their infancy and the state-of-the art of such models is described. Beyond the relationship between amino acid substitution and static protein structure, protein function, and corresponding organismal fitness, other considerations are also discussed. More complex mutational processes such as insertion and deletion and domain rearrangements and even circular permutations should be evaluated. The role of intrinsically disordered proteins is still controversial, but may be increasingly important to consider. Protein geometry and protein dynamics as a deviation from static considerations of protein structure are also important. Protein expression level is known to be a major determinant of evolutionary rate and several considerations including selection at the mRNA level and the role of interaction specificity are discussed. Lastly, the relationship between modeling and needed high-throughput experimental data as well as experimental examination of protein evolution using ancestral sequence resurrection and in vitro biochemistry are presented, towards an aim of ultimately generating better models for biological inference and prediction.
Rapid microsatellite identification from Illumina paired-end genomic sequencing in two birds and a snake
Castoe TA, Poole AW, de Koning AP, Jones KL, Tomback DF, Oyler-McCance SJ, Fike JA, Lance SL, Streicher JW, Smith EN, Pollock DD
Identification of microsatellites, or simple sequence repeats (SSRs), can be a time-consuming and costly investment requiring enrichment, cloning, and sequencing of candidate loci. Recently, however, high throughput sequencing (with or without prior enrichment for specific SSR loci) has been utilized to identify SSR loci. The direct "Seq-to-SSR" approach has an advantage over enrichment-based strategies in that it does not require a priori selection of particular motifs, or prior knowledge of genomic SSR content. It has been more expensive per SSR locus recovered, however, particularly for genomes with few SSR loci, such as bird genomes. The longer but relatively more expensive 454 reads have been preferred over less expensive Illumina reads. Here, we use Illumina paired-end sequence data to identify potentially amplifiable SSR loci (PALs) from a snake (the Burmese python, Python molurus bivittatus), and directly compare these results to those from 454 data. We also compare the python results to results from Illumina sequencing of two bird genomes (Gunnison Sage-grouse, Centrocercus minimus, and Clark's Nutcracker, Nucifraga columbiana), which have considerably fewer SSRs than the python. We show that direct Illumina Seq-to-SSR can identify and characterize thousands of potentially amplifiable SSR loci for as little as $10 per sample--a fraction of the cost of 454 sequencing. Given that Illumina Seq-to-SSR is effective, inexpensive, and reliable even for species such as birds that have few SSR loci, it seems that there are now few situations for which prior hybridization is justifiable.
Sequencing three crocodilian genomes to illuminate the evolution of archosaurs and amniotes
St John JA, Braun EL, Isberg SR, Miles LG, Chong AY, Gongora J, Dalzell P, Moran C, Bed'hom B, Abzhanov A, Burgess SC, Cooksey AM, Castoe TA, Crawford NG, Densmore LD, Drew JC, Edwards SV, Faircloth BC, Fujita MK, Greenwold MJ, Hoffmann FG, Howard JM, Iguchi T, Janes DE, Khan SY, Kohno S, de Koning AJ, Lance SL, McCarthy FM, McCormack JE, Merchant ME, Peterson DG, Pollock DD, Pourmand N, Raney BJ, Roessler KA, Sanford JR, Sawyer RH, Schmidt CJ, Triplett EW, Tuberville TD, Venegas-Anaya M, Howard JT, Jarvis ED, Guillette LJ Jr, Glenn TC, Green RE, Ray DA.
The International Crocodilian Genomes Working Group (ICGWG) will sequence and assemble the American alligator (Alligator mississippiensis), saltwater crocodile (Crocodylus porosus) and Indian gharial (Gavialis gangeticus) genomes. The status of these projects and our planned analyses are described.
The study of biology is fundamentally different from many other scientific pursuits, such as geology or astrophysics. This difference stems from the ubiquitous questions that arise about function and purpose. These are questions concerning why biological objects operate the way they do: what is the function of a polymerase? What is the role of the immune system? No one, aside from the most dedicated anthropist or interventionist theist, would attempt to determine the purpose of the earth's mantle or the function of a binary star. Amont the sciences, it is only biology in which the details of what an object does can be said to be part of the reason for its existence. This is because the process of evolution is capable of improving an object to better carry out a function; that is, it adapts an object within the constraints of mechanics and history (i.e, what has come before). Thus, the ultimate basis of these biological questions is the process of evolution; generally, the function of an enzyme, cell type, organ, system, or trait is the thing that it does that contributes to the fitness (i.e., reproductive success) of the organism of which it is a part or characteristic. Our investigations cannot escape the simple fact that all things in biology (including ourselves) are, ultimately, the result of an evolutionary process.
The understanding of our evolutionary heritage has a wide range of conceptual, theoretical, and practical applications. First, we are often interested in the evolutionary process because it has specific consequences... Second, by observing not just a single instance of something, but also how it varies within and between populations and speciess, we can learn more about how it works and what is important for maintaining or altering function...Third, we are interested in evidence of new things that are not contained in our current philosophy...Fourth, evolutionary biology is the story of our creation, the basis of who we are and why we are here on this planet...This is where art and science meet, both "incandescently" and "incestuously" [2].
Germline TRAV5D-4 T Cell Receptor Sequence Targets a Primary Insulin Peptide of NOD Mice
M. Nakayama, T.A. Castoe, Sosinowski T, He X, Johnson K, Haskins K, Vignali DA, Gapin L, D. D. Pollock, and G.S. Eisenbarth
There is accumulating evidence that autoimmunity to insulin B chain peptide, amino acids 9-23 (insulin B:9-23), is central to development of autoimmune diabetes of the NOD mouse model. We hypothesized that enhanced susceptibility to autoimmune diabetes is the result of targeting of insulin by a T-cell receptor (TCR) sequence commonly encoded in the germline. In this study, we aimed to demonstrate that a particular V? gene TRAV5D-4 with multiple junction sequences is sufficient to induce anti-islet autoimmunity by studying retrogenic mouse lines expressing ?-chains with different V? TRAV genes. Retrogenic NOD strains expressing V? TRAV5D-4 ?-chains with many different complementarity determining region (CDR) 3 sequences, even those derived from TCRs recognizing islet-irrelevant molecules, developed anti-insulin autoimmunity. Induction of insulin autoantibodies by TRAV5D-4 ?-chains was abrogated by the mutation of insulin peptide B:9-23 or that of two amino acid residues in CDR1 and 2 of the TRAV5D-4. TRAV13-1, the human ortholog of murine TRAV5D-4, was also capable of inducing in vivo anti-insulin autoimmunity when combined with different murine CDR3 sequences. Targeting primary autoantigenic peptides by simple germline-encoded TCR motifs may underlie enhanced susceptibility to the development of autoimmune diabetes.
LTR retrotransposons contribute to genomic gigantism in plethodontid salamanders
Sun C, Shepard DB, Chong RA, López Arriaza J, Hall K, Castoe TA, Feschotte C, Pollock DD, Mueller RL
Among vertebrates, most of the largest genomes are found within the salamanders, a clade of amphibians that includes 613 species. Salamander genome sizes range from ~14 to ~120 Gb. Because genome size is correlated with nucleus and cell sizes, as well as other traits, morphological evolution in salamanders has been profoundly affected by genomic gigantism. However, the molecular mechanisms driving genomic expansion in this clade remain largely unknown. Here, we present the first comparative analysis of transposable element (TE) content in salamanders. Using high-throughput sequencing, we generated genomic shotgun data for six species from the Plethodontidae, the largest family of salamanders. We then developed a pipeline to mine TE sequences from shotgun data in taxa with limited genomic resources, such as salamanders. Our summaries of overall TE abundance and diversity for each species demonstrate that TEs make up a substantial portion of salamander genomes, and that all of the major known types of TEs are represented in salamanders. The most abundant TE superfamilies found in the genomes of our six focal species are similar, despite substantial variation in genome size. However, our results demonstrate a major difference between salamanders and other vertebrates: salamander genomes contain much larger amounts of long terminal repeat (LTR) retrotransposons, primarily Ty3/gypsy elements. Thus, the extreme increase in genome size that occurred in salamanders was likely accompanied by a shift in TE landscape. These results suggest that increased proliferation of LTR retrotransposons was a major molecular mechanism contributing to genomic expansion in salamanders.
Repetitive elements may comprise over two-thirds of the human genome
A. P. J. de Koning, W. Gu, T. A. Castoe, M. A. Batzer, and D. D. Pollock
Transposable elements (TEs) are conventionally identified in eukaryotic genomes by alignment to consensus element sequences. Using this approach, about half of the human genome has been previously identified as TEs and low-complexity repeats. We recently developed a highly sensitive alternative de novo strategy, P-clouds, that instead searches for clusters of high-abundance oligonucleotides that are related in sequence space (oligo "clouds"). We show here that P-clouds predicts >840 Mbp of additional repetitive sequences in the human genome, thus suggesting that 66%-69% of the human genome is repetitive or repeat-derived. To investigate this remarkable difference, we conducted detailed analyses of the ability of both P-clouds and a commonly used conventional approach, RepeatMasker (RM), to detect different sized fragments of the highly abundant human Alu and MIR SINEs. RM can have surprisingly low sensitivity for even moderately long fragments, in contrast to P-clouds, which has good sensitivity down to small fragment sizes (~25 bp). Although short fragments have a high intrinsic probability of being false positives, we performed a probabilistic annotation that reflects this fact. We further developed "element-specific" P-clouds (ESPs) to identify novel Alu and MIR SINE elements, and using it we identified ~100 Mb of previously unannotated human elements. ESP estimates of new MIR sequences are in good agreement with RM-based predictions of the amount that RM missed. These results highlight the need for combined, probabilistic genome annotation approaches and suggest that the human genome consists of substantially more repetitive sequence than previously believed.
Sequencing the genome of the Burmese python (Python molurus bivittatus) as a model for studying extreme adaptations in snakes
T. A. Castoe, A. P. J. de Koning, K. T. Hall, K. D. Yokoyama, W. Gu, E. N. Smith , C. Feschotte, P. Uetz, D. A. Ray, J. Dobry, R. Bogden, S. P. Mackessy, A. M. Bronikowski, W. C. Warren, S. M. Secor, and D. D. Pollock
The Consortium for Snake Genomics is in the process of sequencing the genome and creating transcriptomic resources for the Burmese python. Here, we describe how this will be done, what analyses this work will include, and provide a timeline.
Bayesian analysis of high-throughput quantitative measurement of protein-DNA interactions
Pollock, D. D, A. P. J. de Koning, T. A. Castoe, M. E. Churchill, and K. J. Kechris
Transcriptional regulation depends upon the binding of transcription factor (TF) proteins to DNA in a sequence-dependent manner. Although many experimental methods address the interaction between DNA and proteins, they generally do not comprehensively and accurately assess the full binding repertoire (the complete set of sequences that might be bound with at least moderate strength). Here, we develop and evaluate through simulation an experimental approach that allows simultaneous high-throughput quantitative analysis of TF binding affinity to thousands of potential DNA ligands. Tens of thousands of putative binding targets can be mixed with a TF, and both the pre-bound and bound target pools sequenced. A hierarchical Bayesian Markov chain Monte Carlo approach determines posterior estimates for the dissociation constants, sequence-specific binding energies, and free TF concentrations. A unique feature of our approach is that dissociation constants are jointly estimated from their inferred degree of binding and from a model of binding energetics, depending on how many sequence reads are available and the explanatory power of the energy model. Careful experimental design is necessary to obtain accurate results over a wide range of dissociation constants. This approach, which we call Simultaneous Ultra high-throughput Ligand Dissociation EXperiment (SULDEX), is theoretically capable of rapid and accurate elucidation of an entire TF-binding repertoire.
A multi-organ transcriptome resource for the Burmese Python (Python molurus bivittatus)
Castoe, T. A., S. E. Fox, A. P. J. de Koning, A. W. Poole, J. M. Daza, E. N. Smith, T. C. Mockler, S. M Secor, and D. D. Pollock
BACKGROUND:
Snakes provide a unique vertebrate system for studying a diversity of extreme adaptations, including those related to development, metabolism, physiology, and venom. Despite their importance as research models, genomic resources for snakes are few. Among snakes, the Burmese python is the premier model for studying extremes of metabolic fluctuation and physiological remodelling. In this species, the consumption of large infrequent meals can induce a 40-fold increase in metabolic rate and more than a doubling in size of some organs. To provide a foundation for research utilizing the python, our aim was to assemble and annotate a transcriptome reference from the heart and liver. To accomplish this aim, we used the 454-FLX sequencing platform to collect sequence data from multiple cDNA libraries.
RESULTS:
We collected nearly 1 million 454 sequence reads, and assembled these into 37,245 contigs with a combined length of 13,409,006 bp. To identify known genes, these contigs were compared to chicken and lizard gene sets, and to all Genbank sequences. A total of 13,286 of these contigs were annotated based on similarity to known genes or Genbank sequences. We used gene ontology (GO) assignments to characterize the types of genes in this transcriptome resource. The raw data, transcript contig assembly, and transcript annotations are made available online for use by the broader research community.
CONCLUSION:
These data should facilitate future studies using pythons and snakes in general, helping to further contribute to the utilization of snakes as a model evolutionary and physiological system. This sequence collection represents a major genomic resource for the Burmese python, and the large number of transcript sequences characterized should contribute to future research in this and other snake species.
the evolution of venom repertoires.
Discovery of highly divergent repeat landscapes in snake genomes using high throughput sequencing
Castoe, T. A., K. Hall, M. L. Guibotsy Mboulas, W. Gu, A. P. J. de Koning, A. W. Poole, V. Vemulapalli, J. M. Daza, C. Feschotte, and D. D. Pollock
We conducted a comprehensive assessment of genomic repeat content in two snake genomes, the venomous copperhead (Agkistrodon contortrix) and the Burmese python (Python molurus bivittatus). These two genomes are both relatively small (~1.4 Gb), but have surprisingly extensive differences in the abundance and expansion histories of their repeat elements. In the python, the readily identifiable repeat element content is low (21%), similar to bird genomes, whereas that of the copperhead is higher (45%), similar to mammalian genomes. The copperhead's greater repeat content arises from the recent expansion of many different microsatellites and TE families, and the copperhead had 23-fold greater levels of TE-related transcripts than the python. This suggests the possibility that greater TE activity in the copperhead is ongoing. Expansion of CR1 LINEs in the copperhead genome has resulted in TE-mediated microsatellite expansion ("microsatellite seeding") at a scale several orders of magnitude greater than previously observed in vertebrates. Snakes also appear to be prone to horizontal transfer of TEs, particularly in the copperhead lineage. The reason that the copperhead has such a small genome in the face of so much recent expansion of repeat elements remains an open question, although selective pressure related to extreme metabolic performance is an obvious candidate. TE activity can affect gene regulation as well as rates of recombination and gene duplication, and it is therefore possible that TE activity played a role in the evolution of major adaptations in snakes; some evidence suggests this may include the evolution of venom repertoires.
The genome of the green anole lizard and a comparative analysis with birds and mammals
Alföldi, J., , T.A. Castoe,..., D.D Pollock, ..., K. Linblad-Toh
The evolution of the amniotic egg was one of the great evolutionary innovations in the history of life, freeing vertebrates from an obligatory connection to water and thus permitting the conquest of terrestrial environments. Among amniotes, genome sequences are available for mammals and birds, but not for non-avian reptiles. Here we report the genome sequence of the North American green anole lizard, Anolis carolinensis. We find that A. carolinensis microchromosomes are highly syntenic with chicken microchromosomes, yet do not exhibit the high GC and low repeat content that are characteristic of avian microchromosomes. Also, A. carolinensis mobile elements are very young and diverse-more so than in any other sequenced amniote genome. The GC content of this lizard genome is also unusual in its homogeneity, unlike the regionally variable GC content found in mammals and birds. We describe and assign sequence to the previously unknown A. carolinensis X chromosome. Comparative gene analysis shows that amniote egg proteins have evolved significantly more rapidly than other proteins. An anole phylogeny resolves basal branches to illuminate the history of their repeated adaptive radiations.
A proposal to sequence the genome of a garter snake
Castoe, T.A., A.M. Bronikowski, E.D. Brodie III, S.V. Edwards, M.E. Pfrender, M.D. Shipiro, D.D. Pollock, and W.C. Warren
Here we develop an argument in support of sequencing a garter snake (Thamnophis sirtalis) genome, and outline a plan to accomplish this. This snake is a common, widespread, nonvenomous North American species that has served as a model for diverse studies in evolutionary biology, physiology, genomics, behavior and coevolution. The anole lizard is currently the lone whole-genome sequence available for a non-avian reptile. Thus, the garter snake would be the first available snake genome sequence and as such would provide much needed comparative representation of non-avian reptilian genomes, and would also allow critical new insights about vertebrate comparative genomics studies in general. We outline the major areas of discovery that the availability of the garter snake genome would enable, and describe a plan for whole-genome sequencing.
Prior to the availability of multiple eukaryotic genomes, it was expected that innovation and divergence at the phenotypic level would be readily explained by molecular innovation and divergence in protein-coding genes. Thus far, however, evidence for adaptation in proteins as a causative explanation of organismal diversity is rare, particularly in the vertebrates. While it may be unreasonable to expect to explain the origins of all phenotypic diversity through adaptation of proteins, it is only reasonable to assume that we have missed an extremely large number of such cases. Given the tremendous acceleration of genome biology enabled by next-generation sequencing, we must revisit this question and ask ourselves what we may intuitively expect and how we can reasonably search for it. This chapter represents our perspective on how this may be achieved
Warren WC, Clayton DF, Ellegren H, Arnold AP, Hillier LW, Künstner A, Searle S, White S, Vilella AJ, Fairley S, Heger A, Kong L, Ponting CP, Jarvis ED, Mello CV, Minx P, Lovell P, Velho TA, Ferris M, Balakrishnan CN, Sinha S, Blatti C, London SE, Li Y, Lin YC, George J, Sweedler J, Southey B, Gunaratne P, Watson M, Nam K, Backström N, Smeds L, Nabholz B, Itoh Y, Whitney O, Pfenning AR, Howard J, Völker M, Skinner BM, Griffin DK, Ye L, McLaren WM, Flicek P, Quesada V, Velasco G, Lopez-Otin C, Puente XS, Olender T, Lancet D, Smit AF, Hubley R, Konkel MK, Walker JA, Batzer MA, Gu W, Pollock DD, Chen L, Cheng Z, Eichler EE, Stapley J, Slate J, Ekblom R, Birkhead T, Burke T, Burt D, Scharff C, Adam I, Richard H, Sultan M, Soldatov A, Lehrach H, Edwards SV, Yang SP, Li X, Graves T, Fulton L, Nelson J, Chinwalla A, Hou S, Mardis ER, Wilson RK
The zebra finch is an important model organism in several fields with unique relevance to human neuroscience. Like other songbirds, the zebra finch communicates through learned vocalizations, an ability otherwise documented only in humans and a few other animals and lacking in the chicken-the only bird with a sequenced genome until now. Here we present a structural, functional and comparative analysis of the genome sequence of the zebra finch (Taeniopygia guttata), which is a songbird belonging to the large avian order Passeriformes. We find that the overall structures of the genomes are similar in zebra finch and chicken, but they differ in many intrachromosomal rearrangements, lineage-specific gene family expansions, the number of long-terminal-repeat-based retrotransposons, and mechanisms of sex chromosome dosage compensation. We show that song behaviour engages gene regulatory networks in the zebra finch brain, altering the expression of long non-coding RNAs, microRNAs, transcription factors and their targets. We also show evidence for rapid molecular evolution in the songbird lineage of genes that are regulated during song experience. These results indicate an active involvement of the genome in neural processes underlying vocal communication and identify potential genetic substrates for the evolution and regulation of this behaviour.
Comparison of normalization methods for construction of large multiplex amplicon pools for next-generation sequencing
J. K. Harris, J.W. Sahl, T.A. Castoe, D.D. Pollock, and J.R. Spear
Constructing mixtures of tagged or bar-coded DNAs for sequencing is an important requirement for the efficient use of next-generation sequencers in applications where limited sequence data are required per sample. There are many applications in which next-generation sequencing can be used effectively to sequence large mixed samples; an example is the characterization of microbial communities where 1,000 sequences per samples are adequate to address research questions. Thus, it is possible to examine hundreds to thousands of samples per run on massively parallel next-generation sequencers. However, the cost savings for efficient utilization of sequence capacity is realized only if the production and management costs associated with construction of multiplex pools are also scalable. One critical step in multiplex pool construction is the normalization process, whereby equimolar amounts of each amplicon are mixed. Here we compare three approaches (spectroscopy, size-restricted spectroscopy, and quantitative binding) for normalization of large, multiplex amplicon pools for performance and efficiency. We found that the quantitative binding approach was superior and represents an efficient scalable process for construction of very large, multiplex pools with hundreds and perhaps thousands of individual amplicons included. We demonstrate the increased sequence diversity identified with higher throughput. Massively parallel sequencing can dramatically accelerate microbial ecology studies by allowing appropriate replication of sequence acquisition to account for temporal and spatial variations. Further, population studies to examine genetic variation, which require even lower levels of sequencing, should be possible where thousands of individual bar-coded amplicons are examined in parallel.
Gene-specific RNA polymerase II phosphorylation and the CTD code
H. Kim, B. Erickson, W. Luo, D. Seward, J. H. Graber, D.D. Pollock, P. C. Megee, and D. L. Bentley
Phosphorylation of the RNA polymerase (Pol) II C-terminal domain (CTD) repeats (1-YSPTSPS-7) is coupled to transcription and may act as a 'code' that controls mRNA synthesis and processing. To examine the code in budding yeast, we mapped genome-wide CTD Ser2, Ser5 and Ser7 phosphorylations and the CTD-associated termination factors Nrd1 and Pcf11. Phospho-CTD dynamics are not scaled to gene length and are gene-specific, with highest Ser5 and Ser7 phosphorylation at the 5' ends of well-expressed genes with nucleosome-occupied promoters. The CTD kinases Kin28 and Ctk1 markedly affect Pol II distribution in a gene-specific way. The code is therefore written differently on different genes, probably under the control of promoters. Ser7 phosphorylation is enriched on introns and at sites of Nrd1 accumulation, suggesting links to splicing and Nrd1 recruitment. Nrd1 and Pcf11 frequently colocalize, suggesting functional overlap. Unexpectedly, Pcf11 is enriched at centromeres and Pol III-transcribed genes.
Rapid likelihood analysis on large phylogenies using partial sampling of substitution histories
A. P. J. de Koning, W. Gu, and D. D. Pollock
The strength and pattern of coevolution between amino acid residues varies depending on their structural and functional environment. This context dependence, along with differences in analytical technique, is responsible for different results among coevolutionary analyses of different proteins. It is thus important to perform detailed study of individual proteins to gain better insight into how context dependence can affect coevolutionary patterns even within individual proteins, and to unravel the details of context dependence with respect to structure and function. Here, we extend our previous study by presenting further analysis of residue coevolution in cytochrome c oxidase subunit I sequences from 231 vertebrates using a statistically robust phylogeny-based maximum likelihood ratio method. As in previous studies, a strong overall coevolutionary signal was detected, and coevolution within structural regions was significantly related to the Ca distances between residues. While the strong selection for adjacent residues among predicted coevolving pairs in the surface region indicates that the statistical method is highly selective for biologically relevant interactions, the coevolutionary signal was strongest in the transmembrane region, although the distances between coevolving residues were greater. This indicates that coevolution may act to maintain more global structural and functional constraints in the transmembrane region. In the transmembrane region, sites that coevolved according to polarity and hydrophobicity rather than volume had a greater tendency to co-localize with just one of the predicted proton channels (channel H). Thus, the details of coevolution in cytochrome c oxidase subunit I depend greatly on domain structure and residue physicochemical characteristics, but proximity to function appears to play a critical role. We hypothesize that the association of coevolutionary sites with channel H was caused by adaptive coevolution, and is indicative of a more important functional role for this channel.
Adaptive molecular convergencesMolecular evolution versus molecular phylogenetics
T. A. Castoe*, A. P. J. de Koning*, and D. D. Pollock
The strength and pattern of coevolution between amino acid residues varies depending on their structural and functional environment. This context dependence, along with differences in analytical technique, is responsible for different results among coevolutionary analyses of different proteins. It is thus important to perform detailed study of individual proteins to gain better insight into how context dependence can affect coevolutionary patterns even within individual proteins, and to unravel the details of context dependence with respect to structure and function. Here, we extend our previous study by presenting further analysis of residue coevolution in cytochrome c oxidase subunit I sequences from 231 vertebrates using a statistically robust phylogeny-based maximum likelihood ratio method. As in previous studies, a strong overall coevolutionary signal was detected, and coevolution within structural regions was significantly related to the Ca distances between residues. While the strong selection for adjacent residues among predicted coevolving pairs in the surface region indicates that the statistical method is highly selective for biologically relevant interactions, the coevolutionary signal was strongest in the transmembrane region, although the distances between coevolving residues were greater. This indicates that coevolution may act to maintain more global structural and functional constraints in the transmembrane region. In the transmembrane region, sites that coevolved according to polarity and hydrophobicity rather than volume had a greater tendency to co-localize with just one of the predicted proton channels (channel H). Thus, the details of coevolution in cytochrome c oxidase subunit I depend greatly on domain structure and residue physicochemical characteristics, but proximity to function appears to play a critical role. We hypothesize that the association of coevolutionary sites with channel H was caused by adaptive coevolution, and is indicative of a more important functional role for this channel.
Rapid identification of thousands of copperhead snake (Agkistrodon contortrix) microsatellite loci from modest amounts of 454 shotgun genome sequence
T. A. Castoe, A. W. Poole, W. Gu, A. P. J. de Koning, J. M. Daza, E. N. Smith, and D. D. Pollock
Optimal integration of next-generation sequencing into mainstream research requires re-evaluation of how problems can be reasonably overcome and what questions can be asked. One potential application is the rapid acquisition of genomic information to identify microsatellite loci for evolutionary, population genetic and chromosome linkage mapping research on non-model and not previously sequenced organisms. Here, we report on results using high-throughput sequencing to obtain a large number of microsatellite loci from the venomous snake Agkistrodon contortrix, the copperhead. We used the 454 Genome Sequencer FLX next-generation sequencing platform to sample randomly ~27 Mbp (128 773 reads) of the copperhead genome, thus sampling about 2% of the genome of this species. We identified microsatellite loci in 11.3% of all reads obtained, with 14 612 microsatellite loci identified in total, 4564 of which had flanking sequences suitable for polymerase chain reaction primer design. The random sequencing-based approach to identify microsatellites was rapid, cost-effective and identified thousands of useful microsatellite loci in a previously unstudied species.
From the Cover: Evidence for an ancient adaptive episode of convergent molecular evolution
T. A. Castoe*, A. P. J. de Koning*, H. Kim, W. Gu, B. P Noonan, G. Naylor, Z. J. Jiang, C. L. Parkinson, and D. D. Pollock
Documented cases of convergent molecular evolution due to selection are fairly unusual, and examples to date have involved only a few amino acid positions. However, because convergence mimics shared ancestry and is not accommodated by current phylogenetic methods, it can strongly mislead phylogenetic inference when it does occur. Here, we present a case of extensive convergent molecular evolution between snake and agamid lizard mitochondrial genomes that overcomes an otherwise strong phylogenetic signal. Evidence from morphology, nuclear genes, and most sites in the mitochondrial genome support one phylogenetic tree, but a subset of mostly amino acid-altering substitutions (primarily at the first and second codon positions) across multiple mitochondrial genes strongly supports a radically different phylogeny. The relevant sites generally evolved slowly but converged between ancient lineages of snakes and agamids. We estimate that approximately 44 of 113 predicted convergent changes distributed across all 13 mitochondrial protein-coding genes are expected to have arisen from nonneutral causes-a remarkably large number. Combined with strong previous evidence for adaptive evolution in snake mitochondrial proteins, it is likely that much of this convergent evolution was driven by adaptation. These results indicate that nonneutral convergent molecular evolution in mitochondria can occur at a scale and intensity far beyond what has been documented previously, and they highlight the vulnerability of standard phylogenetic methods to the presence of nonneutral convergent sequence evolution.
Dynamic nucleotide mutation gradients and control region usage in squamate reptile mitochondrial genomes
T. A. Castoe, W. Gu, A. P. J. de Koning, J. M. Gaza, H. Kim, Z. J. Jiang, C. L. Parkinson, and D. D. Pollock
Gradients of nucleotide bias and substitution rates occur in vertebrate mitochondrial genomes due to the asymmetric nature of the replication process. The evolution of these gradients has previously been studied in detail in primates, but not in other vertebrate groups. From the primate study, the strengths of these gradients are known to evolve in ways that can substantially alter the substitution process, but it is unclear how rapidly they evolve over evolutionary time or how different they may be in different lineages or groups of vertebrates. Given the importance of mitochondrial genomes in phylogenetics and molecular evolutionary research, a better understanding of how asymmetric mitochondrial substitution gradients evolve would contribute key insights into how this gradient evolution may mislead evolutionary inferences, and how it may also be incorporated into new evolutionary models. Most snake mitochondrial genomes have an additional interesting feature, 2 nearly identical control regions, which vary among different species in the extent that they are used as origins of replication. Given the expanded sampling of complete snake genomes currently available, together with 2 additional snakes sequenced in this study, we reexamined gradient strength and CR usage in alethinophidian snakes as well as several lizards that possess dual CRs. Our results suggest that nucleotide substitution gradients (and corresponding nucleotide bias) and CR usage is highly labile over the approximately 200 m.y. of squamate evolution, and demonstrates greater overall variability than previously shown in primates. The evidence for the existence of such gradients, and their ability to evolve rapidly and converge among unrelated species suggests that gradient dynamics could easily mislead phylogenetic and molecular evolutionary inferences, and argues strongly that these dynamics should be incorporated into phylogenetic models.
Identifying DNA strands using a kernel of classified sequences
Tonnsman, G., D. D. Pollock, W. Gu, and T. A. Castoe
Automated DNA sequencing produces a large amount of raw DNA sequence data that then needs to be classified, organized, and annotated. One major application is the comparison of new DNA sequences with previously known classified sequences. In this paper we present a new approach to perform these comparisons. From a kernel of previously classified DNA sequences, we identify distinctive oligomers, or short DNA sequences, that are infrequent and thus highly unique within the kernel. We then search for the presence of these distinctive oligomers in the new unclassified DNA sequences. Their presence indicates a possible relation between a new DNA sequence and every previously classified DNA sequence that shares the distinctive oligomer. Ultimately, unclassified sequences are related to classified sequences with which they share the highest number of distinctive oligomers. We explain the details of our technique and show some experimental results in a kernel of immunoglobulin DNA sequences.
Intrinsic amino acid side-chain hydrophilicity/hydrophobicity coefficients determined by reversed-phase high-performance liquid chromatography of model peptides: Comparison with other hydrophilicity/hydrophobicity scales
C. T. Mant, J. M. Kovacs, H. Kim, and D. D. Pollock, and R.S. Hodges
An accurate determination of the intrinsic hydrophilicity/hydrophobicity of amino acid side-chains in peptides and proteins is fundamental in understanding many area of research, including protein folding and stability, peptide and protein function, protein-protein interactions and peptide/protein oligomerization, as well as the design of protocols for purification and characterization of peptides and proteins. Our definition of intrinsic hydrophilicity/hydrophobicity of side-chains is the maximum possible hydrophilicity/hydrophobicity of side-chains in the absence of any nearest-neighbor effects and/or any conformational effects of the polypeptide chain that prevent full expression of side-chain hydrophilicity/hydrophobicity. In this review, we have compared an experimentally derived intrinsic side-chain hydrophilicity/hydrophobicity scale generated from RP-HPLC retention behavior of de novo designed synthetic model peptides at pH 2 and pH 7 with other RP-HPLC-derived scales, as well as scales generated from classic experimental and calculation-based methods of octanol/water partitioning of Nalpha-acetyl-amino-acid amides or free energy of transfer of free amino acids. Generally poor correlation was found with previous RP-HPLC-derived scales, likely due to the random nature of the peptide mixtures in terms of varying peptide size, conformation and frequency of particular amino acids. In addition, generally poor correlation with the classical approaches served to underline the importance of the presence of a polypeptide backbone when generating intrinsic values. We have shown that the intrinsic scale determined here is in full agreement with the structural characteristics of amino acid side-chains.
Adaptive evolution and functional redesign of core metabolic proteins in snakes
T. A. Castoe, Z. J. Jiang, Z. O. Wang, W. Gu, and D. D. Pollock
BACKGROUND:
Adaptive evolutionary episodes in core metabolic proteins are uncommon, and are even more rarely linked to major macroevolutionary shifts.
METHODOLOGY/PRINCIPAL FINDINGS:
We conducted extensive molecular evolutionary analyses on snake mitochondrial proteins and discovered multiple lines of evidence suggesting that the proteins at the core of aerobic metabolism in snakes have undergone remarkably large episodic bursts of adaptive change. We show that snake mitochondrial proteins experienced unprecedented levels of positive selection, coevolution, convergence, and reversion at functionally critical residues. We examined Cytochrome C oxidase subunit I (COI) in detail, and show that it experienced extensive modification of normally conserved residues involved in proton transport and delivery of electrons and oxygen. Thus, adaptive changes likely altered the flow of protons and other aspects of function in CO, thereby influencing fundamental characteristics of aerobic metabolism. We refer to these processes as "evolutionary redesign" because of the magnitude of the episodic bursts and the degree to which they affected core functional residues.
CONCLUSIONS/SIGNIFICANCE:
The evolutionary redesign of snake COI coincided with adaptive bursts in other mitochondrial proteins and substantial changes in mitochondrial genome structure. It also generally coincided with or preceded major shifts in ecological niche and the evolution of extensive physiological adaptations related to lung reduction, large prey consumption, and venom evolution. The parallel timing of these major evolutionary events suggests that evolutionary redesign of metabolic and mitochondrial function may be related to, or underlie, the extreme changes in physiological and metabolic efficiency, flexibility, and innovation observed in snake evolution.
Identification of repeat structure in large genomes using repeat probability clouds
W. Gu, T. A. Castoe, D. J. Hedges, M. A. Batzer, and D. D. Pollock
The identification of repeat structure in eukaryotic genomes can be time-consuming and difficult because of the large amount of information ( approximately 3 x 10(9) bp) that needs to be processed and compared. We introduce a new approach based on exact word counts to evaluate, de novo, the repeat structure present within large eukaryotic genomes. This approach avoids sequence alignment and similarity search, two of the most time-consuming components of traditional methods for repeat identification. Algorithms were implemented to efficiently calculate exact counts for any length oligonucleotide in large genomes. Based on these oligonucleotide counts, oligonucleotide excess probability clouds, or "P-clouds," were constructed. P-clouds are composed of clusters of related oligonucleotides that occur, as a group, more often than expected by chance. After construction, P-clouds were mapped back onto the genome, and regions of high P-cloud density were identified as repetitive regions based on a sliding window approach. This efficient method is capable of analyzing the repeat content of the entire human genome on a single desktop computer in less than half a day, at least 10-fold faster than current approaches. The predicted repetitive regions strongly overlap with known repeat elements as well as other repetitive regions such as gene families, pseudogenes, and segmental duplicons. This method should be extremely useful as a tool for use in de novo identification of repeat structure in large newly sequenced genomes.
Structural, biochemical, and in vivo characterization of the first virally encoded cyclophilin from the Mimivirus
Thai V, Renesto P, Fowler A, Brown D, Davis T, Gu W, Pollock DD, Kern D, Raoult D, and Eisenmesser E
Although multiple viruses utilize host cell cyclophilins, including SARS and HIV-1, their role in infection is poorly understood. To help elucidate these roles, we have characterized the first virally encoded cyclophilin (mimicyp) derived from the largest virus discovered to date (the Mimivirus) that is also a causative agent of pneumonia in humans. Mimicyp adopts a typical cyclophilin-fold, yet it also forms trimers unlike any
previously characterized homologue. Strikingly, immunofluorescence assays reveal that
mimicyp localizes to the surface of the mature virion, as recently proposed for several
viruses that recruit host cell cyclophilins such as SARS and HIV-1. Additionally mimicyp
lacks peptidyl-prolyl isomerase activity in contrast to human cyclophilins. Thus, this study
suggests that cyclophilins, whether recruited from host cells (i.e. HIV-1 and SARS) or
virally encoded (i.e. Mimivirus), are localized on viral surfaces for at least a subset of
viruses.
Phylogenomics, protein family evolution, and the Tree of Life: an integrated approach between molecular evolution and computational intelligence
Naihum LA and Pereira SL
The massive amount of information generated by genomic technologies has opened new frontiers in science by bridging disciplines such as computational biology, molecular biology, molecular evolution, evolutionary biology, and ecology. Many tools and methods have been developed over the past several years to allow analysis of molecular sequences. Phylogenomics, the interpretation of genomic data to determine gene function and phylogenetic relationships of organisms, remains challenging nevertheless. Here, we focus on the application of phylogenomics to improve functional prediction of genes/products, to understand the evolution of protein families, and to resolve phylogenetic relationships of organisms. We point out areas that require further development, such as computational tools and methods to manipulate large and diverse data sets. The application of an integrated computational and biological approach may help to achieve a better system-based understanding of biological processes in different environments. This will help to fully access valuable information available from the evolution of genes, and genomes in the wide diversity of intact organisms and biological communities.
Coevolutionary patterns in cytochrome c oxidase
subunit I depend on structure and functional context
Wang ZO and Pollock DD
The strength and pattern of coevolution between amino acid residues varies depending on their structural and functional environment. This context dependence, along with differences in analytical technique, is responsible for different results among coevolutionary analyses of different proteins. It is thus important to perform detailed study of individual proteins to gain better insight into how context dependence can affect coevolutionary patterns even within individual proteins, and to unravel the details of context dependence with respect to structure and function. Here, we extend our previous study by presenting further analysis of residue coevolution in cytochrome c oxidase subunit I sequences from 231 vertebrates using a statistically robust phylogeny-based maximum likelihood ratio method. As in previous studies, a strong overall coevolutionary signal was detected, and coevolution within structural regions was significantly related to the Ca distances between residues. While the strong selection for adjacent residues among predicted coevolving pairs in the surface region indicates that the statistical method is highly selective for biologically relevant interactions, the coevolutionary signal was strongest in the transmembrane region, although the distances between coevolving residues were greater. This indicates that coevolution may act to maintain more global structural and functional constraints in the transmembrane region. In the transmembrane region, sites that coevolved according to polarity and hydrophobicity rather than volume had a greater tendency to co-localize with just one of the predicted proton channels (channel H). Thus, the details of coevolution in cytochrome c oxidase subunit I depend greatly on domain structure and residue physicochemical characteristics, but proximity to function appears to play a critical role. We hypothesize that the association of coevolutionary sites with channel H was caused by adaptive coevolution, and is indicative of a more important functional role for this channel.
BACKGROUND: The mitochondrial genomes of snakes are characterized by an overall evolutionary rate that appears to be one of the most accelerated among vertebrates. They also possess other unusual features, including short tRNAs and other genes, and a duplicated control region that has been stably maintained since it originated more than 70 million years ago. Here, we provide a detailed analysis of evolutionary dynamics in snake mitochondrial genomes to better understand the basis of these extreme characteristics, and to explore the relationship between mitochondrial genome molecular evolution, genome architecture, and molecular function. We sequenced complete mitochondrial genomes from Slowinski's corn snake (Pantherophis slowinskii) and two cottonmouths (Agkistrodon piscivorus) to complement previously existing mitochondrial genomes, and to provide an improved comparative view of how genome architecture affects molecular evolution at contrasting levels of divergence. RESULTS: We present a Bayesian genetic approach that suggests that the duplicated control region can function as an additional origin of heavy strand replication. The two control regions also appear to have different intra-specific versus inter-specific evolutionary dynamics that may be associated with complex modes of concerted evolution. We find that different genomic regions have experienced substantial accelerated evolution along early branches in snakes, with different genes having experienced dramatic accelerations along specific branches. Some of these accelerations appear to coincide with, or subsequent to, the shortening of various mitochondrial genes and the duplication of the control region and flanking tRNAs. CONCLUSION: Fluctuations in the strength and pattern of selection during snake evolution have had widely varying gene-specific effects on substitution rates, and these rate accelerations may have been functionally related to unusual changes in genomic architecture. The among-lineage and among-gene variation in rate dynamics observed in snakes is the most extreme thus far observed in animal genomes, and provides an important study system for further evaluating the biochemical and physiological basis of evolutionary pressures in vertebrate mitochondria.
Genome of the marsupial Monodelphis domestica reveals innovation in non-coding sequences
Mikkelsen TS, Wakefield MJ, Aken B, Amemiya CT, Chang JL, Duke S, Garber M, Gentles AJ, Goodstadt L, Heger A, Jurka J, Kamal M, Mauceli E, Searle SM, Sharpe T, Baker ML, Batzer MA, Benos PV, Belov K, Clamp M, Cook A, Cuff J, Das R, Davidow L, Deakin JE, Fazzari MJ, Glass JL, Grabherr M, Greally JM, Gu W, Hore TA, Huttley GA, Kleber M, Jirtle RL, Koina E, Lee JT, Mahony S, Marra MA, Miller RD, Nicholls RD, Oda M, Papenfuss AT, Parra ZE, Pollock DD, Ray DA, Schein JE, Speed TP, Thompson K, VandeBerg JL, Wade CM, Walker JA, Waters PD, Webber C, Weidman JR, Xie X, Zody MC; Broad Institute Genome Sequencing Platform, Broad Institute Whole Genome Assembly Team, Broad Institute Whole Genome Assembly Team, Jaffe DB, Alvarez P, Brockman W, Butler J, Chin C, Gnerre S, MacCallum I, Graves JA, Ponting CP, Breen M, Samollow PB, Lander ES, and Lindblad-Toh K
We report a high-quality draft of the genome sequence of the grey, short-tailed opossum (Monodelphis domestica). As the first metatherian ('marsupial') species to be sequenced, the opossum provides a unique perspective on the organization and evolution of mammalian genomes. Distinctive features of the opossum chromosomes provide support for recent theories about genome evolution and function, including a strong influence of biased gene conversion on nucleotide sequence composition, and a relationship between chromosomal characteristics and X chromosome inactivation. Comparison of opossum and eutherian genomes also reveals a sharp difference in evolutionary innovation between protein-coding and non-coding functional elements. True innovation in protein-coding genes seems to be relatively rare, with lineage-specific differences being largely due to diversification and rapid turnover in gene families involved in environmental interactions. In contrast, about 20% of eutherian conserved non-coding elements (CNEs) are recent inventions that postdate the divergence of Eutheria and Metatheria. A substantial proportion of these eutherian-specific CNEs arose from sequence inserted by transposable elements, pointing to transposons as a major creative force in the evolution of mammalian gene regulation.
Evolutionary dynamics of transposable elements in the short-tailed opossum Monodelphis domestica
Gentles AJ, Wakefield MJ, Kohany O, Gu W, Batzer MA, Pollock DD, and Jurka J
The genome of the gray short-tailed opossum Monodelphis domestica is notable for its large size ( approximately 3.6 Gb). We characterized nearly 500 families of interspersed repeats from the Monodelphis. They cover approximately 52% of the genome, higher than in any other amniotic lineage studied to date, and may account for the unusually large genome size. In comparison to other mammals, Monodelphis is significantly rich in non-LTR retrotransposons from the LINE-1, CR1, and RTE families, with >29% of the genome sequence comprised of copies of these elements. Monodelphis has at least four families of RTE, and we report support for horizontal transfer of this non-LTR retrotransposon. In addition to short interspersed elements (SINEs) mobilized by L1, we found several families of SINEs that appear to use RTE elements for mobilization. In contrast to L1-mobilized SINEs, the RTE-mobilized SINEs in Monodelphis appear to shift from G+C-rich to G+C-low regions with time. Endogenous retroviruses have colonized approximately 10% of the opossum genome. We found that their density is enhanced in centromeric and/or telomeric regions of most Monodelphis chromosomes. We identified 83 new families of ancient repeats that are highly conserved across amniotic lineages, including 14 LINE-derived repeats; and a novel SINE element, MER131, that may have been exapted as a highly conserved functional noncoding RNA, and whose emergence dates back to approximately 300 million years ago. Many of these conserved repeats are also present in human, and are highly over-represented in predicted cis-regulatory modules. Seventy-six of the 83 families are present in chicken in addition to mammals.
Regional variation in the density of essential genes in mice
Hentges KE, Pollock DD, Liu B, and Justice MJ
In most species, and particularly in vertebrates, the percentage of genes absolutely required for survival, the essential genes, has not been estimated. To obtain this estimation, we used the mouse as an experimental model to carry out high-efficiency N-ethyl-N-nitrosourea (ENU) mutagenesis screens in two balancer chromosome regions, and compared our results to a third previously published screen. The number of essential genes in each region was predicted based on allele frequencies. We determined that the density of essential genes differs by up to an order of magnitude among genomic regions. This indicates that extrapolating from regional estimates to genome-wide estimates of essential genes has a huge variance. A particularly high density of essential genes on mouse Chromosome 11 coincides with a high degree of regional linkage conservation, providing a possible causal explanation for the density variation. This is the first demonstration of regional variation in essential gene density in the mouse genome.
Evolutionary dynamics of transposable elements in the short-tailed opossum Monodelphis domestica
Gu W, Ray DA, Walker JA, Barnes EW, Gentles AJ, Samollow PB, Jurka J, Batzer MA, and Pollock DD
Short INterspersed Elements (SINEs) are non-autonomous retrotransposons, usually between 100 and 500 base pairs (bp) in length, which are ubiquitous components of eukaryotic genomes. Their activity, distribution, and evolution can be highly informative on genomic structure and evolutionary processes. To determine recent activity, we amplified more than one hundred SINE1 loci in a panel of 43 M. domestica individuals derived from five diverse geographic locations. The SINE1 family has expanded recently enough that many loci were polymorphic, and the SINE1 insertion-based genetic distances among populations reflected geographic distance. Genome-wide comparisons of SINE1 densities and GC content revealed that high SINE1 density is associated with high GC content in a few long and many short spans. Young SINE1s, whether fixed or polymorphic, showed an unbiased GC content preference for insertion, indicating that the GC preference accumulates over long time periods, possibly in periodic bursts. SINE1 evolution is thus broadly similar to human Alu evolution, although it has an independent origin. High GC content adjacent to SINE1s is strongly correlated with bias towards higher AT to GC substitutions and lower GC to AT substitutions. This is consistent with biased gene conversion, and also indicates that like chickens, but unlike eutherian mammals, GC content heterogeneity (isochore structure) is reinforced by substitution processes in the M. domestica genome. Nevertheless, both high and low GC content regions are apparently headed towards lower GC content equilibria, possibly due to a relative shift to lower recombination rates in the recent Monodelphis ancestral lineage. Like eutherians, metatherian (marsupial) mammals have evolved high CpG substitution rates, but this is apparently a convergence in process rather than a shared ancestral state.
Dealing with Uncertainty in Ancestral Sequence Reconstruction: Sampling from the Posterior Distribution
Pollock DD and Chang BS
Resurrection of ancestral proteins in the laboratory to investigate
aspects of their function has provided an exciting opportunity
to experimentally test theories concerning the evolution of
protein structure and function. A potentially important pitfall
of this approach, however, is that sequence and functional bias
in ancestral reconstruction may affect results. In the worst-case
scenario, the bias in reconstruction could lead to incorrect
functional interpretation for reconstructed proteins. Inferring
function or stability based on a single resurrected protein
sequence may be a risky proposition without concurrent examination
to determine if a bias in functional shifts indeed exists. If
the evolutionary process can be modeled fairly well, an effective
means to eliminate the reconstruction bias is to sample ancestral
proteins from the posterior probability space. It is also important
to incorporate uncertainty in the model of evolution and model
variation across sites, and to consider the absence of rare
variants. The question of how many reconstructed ancestral samples
are sufficient to estimate probable ancestral function is an
open one, and it may be specific to the variability in inferred
function among likely ancestors. Given a reasonably accurate
model of evolution, the sampling of even a few proteins from
the posterior may provide a relatively unbiased estimate of
ancestral function, and would allow evaluation of the variance
in this functional estimate. We discuss the details of the problem,
propose a simple experimental approach to solve it, and provide
a program to sample ancestral sequences and to evaluate the
tendency of maximum likelihood estimates to alter amino acid
frequencies and under-sample rare (possibly slightly deleterious)
variants in a protein.
EGenBio: a data management system
for evolutionary genomics and biodiversity
Nahum LA, Reynolds MT, Wang ZO, Faith JJ, Jonna R, Jiang ZJ, Meyer TJ, and Pollock DD
BACKGROUND: Evolutionary genomics requires management and filtering of large numbers of diverse genomic sequences for accurate analysis and inference on evolutionary processes of genomic and functional change. We developed Evolutionary Genomics and Biodiversity (EGenBio; http://egenbio.lsu.edu webcite) to begin to address this. DESCRIPTION: EGenBio is a system for manipulation and filtering of large numbers of sequences, integrating curated sequence alignments and phylogenetic trees, managing evolutionary analyses, and visualizing their output. EGenBio is organized into three conceptual divisions, Evolution, Genomics, and Biodiversity. The Genomics division includes tools for selecting pre-aligned sequences from different genes and species, and for modifying and filtering these alignments for further analysis. Species searches are handled through queries that can be modified based on a tree-based navigation system and saved. The Biodiversity division contains tools for analyzing individual sequences or sequence alignments, whereas the Evolution division contains tools involving phylogenetic trees. Alignments are annotated with analytical results and modification history using our PRAED format. A miscellaneous Tools section and Help framework are also available. EGenBio was developed around our comparative genomic research and a prototype database of mtDNA genomes. It utilizes MySQL-relational databases and dynamic page generation, and calls numerous custom programs. CONCLUSION: EGenBio was designed to serve as a platform for tools and resources to ease combined analysis in evolution, genomics, and biodiversity.
Assessing the accuracy of ancestral protein
reconstruction methods
Williams PD, Pollock DD, Blackburne BP, and Goldstein RA
The phylogenetic inference of ancestral protein sequences is
a powerful technique for the study of molecular evolution, but
any conclusions drawn from such studies are only as good as
the accuracy of the reconstruction method. Every inference method
leads to errors in the ancestral protein sequence, resulting
in potentially-misleading estimates of the ancestral protein’s
properties. To better understand the conditions of the past,
it is important to understand the accuracy of different methods
and how the resulting errors affect the conclusions drawn. The
Maximum Parsimony (MP) and Maximum Likelihood (ML) inference
methods have been shown to misestimate ancestral nucleotide
frequencies, revealing a consistent and incorrect bias, but
little data for proteins exists, partially because of the difficulty
of finding true ancestral sequences for comparison. To assess
the accuracy of ancestral protein reconstruction methods, we
perform computational population evolution simulations featuring
speciation and divergence events using an off-lattice protein
model where fitness depends on the ability to fold into a specified
target structure. As we know the population of sequences at
each step of the simulation, we can compare these known ancestral
sequences and the resulting thermodynamic properties with those
inferred by MP, ML, and Bayesian methods. We find that MP and,
even more so, ML methods overestimate thermostability and that
a Bayesian analysis, although it does not generate the most
accurate sequences, is the most accurate and most unbiased in
terms of resulting protein properties. This suggests that ancestral
reconstruction studies performed using MP and ML may need to
be re-evaluated.
Observations of amino acid gain and loss
during protein evolution are explained by statistical bias
Goldstein RA and Pollock DD
In the scientific literature, and in molecular evolution in
particular, extravagant claims are oftentimes given exceptional
attention. This is true for unusual inferences of relationships
among organisms, dating of organismal divergence times, and
for reconstruction of function and properties of ancestral proteins.
In all of these cases, misuse of statistics and ignorance of
variation can lead to “phylogenetic optimism”, whereby
confidence in the results is vastly overstated and important
sources of bias ignored. As a case in point, the authors of
a recent manuscript in Nature claim to have discovered “universal
trends” of amino acid gain and loss in protein evolution.
Such an inference of convergent evolution in the same direction
in many different taxa should always be treated with extreme
caution, since inferential bias is a likely explanation for
such a trend. Here, we show that the “universal trend”
in amino acid evolution can be explained by a bias in common
methods for inferring evolutionary trends in proteins. Trends
can be more accurately detected using phylogeny-based Bayesian
methods, but the currently available dataset does not contain
sufficient taxa to make definitive assertions, and previous
assertions are almost certainly unfounded. Variation in amino
acid replacement rates among proteins, among positions within
proteins, and over time currently overwhelms our ability to
make sound claims about such trends.
Selective advantage of recombination in evolving
protein populations: A lattice model study
Williams PD, Pollock DD, and Goldstein RA
Recent research has attempted to clarify the contributions of
several mutational processes, such as substitutions or homologous
recombination. Simplistic, tractable protein models, which determine
the compact native structure phenotype from the sequence genotype,
are well-suited to such studies. In this paper, we use a lattice-protein
model to examine the effects of point mutation and homologous
recombination on evolving populations of proteins. We find that
while the majority of mutation and recombination events are
neutral or deleterious, recombination is far more likely to
be beneficial. This results in a faster increase in fitness
during evolution, although the final fitness level is not significantly
changed. This transient advantage provides an evolutionary advantage
to subpopulations that undergo recombination, allowing fixation
of recombination to occur in the population.
Functionality and the evolution of marginal
stability in proteins: inferences from lattice simulations
Williams PD, Pollock DD, and Goldstein RA
It has been known for some time that many proteins are marginally
stable. This has inspired several explanations. Having noted
that the functionality of many enzymes is correlated with subunit
motion, flexibility, or general disorder, some have suggested
that marginally stable proteins should have an evolutionary
advantage over proteins of differing stability. Others have
suggested that stability and functionality are contradictory
qualities, and that selection for both criteria results in marginally
stable proteins, optimised to satisfy the competing design pressures.
While these explanations are plausible, recent research simulating
the evolution of model proteins has shown that selection for
stability, ignoring any aspects of functionality, can result
in marginally stable proteins because of the underlying makeup
of protein sequence-space. We extend this research by simulating
the evolution of proteins, using a computational protein model
that equates functionality with binding and catalysis. In the
model, marginal stability is not required for ligand-binding
functionality and we observe no competing design pressures.
The resulting proteins are marginally stable, again demonstrating
that neutral evolution is sufficient for explaining marginal
stability in observed proteins.
Divergence, recombination,
and retention of functionality during protein evolution
Xu YO, Hall RW, Goldstein RA, Pollock DD.
Protein structure and function are not easily predictable from
primary sequence, and because of this we have only a vague idea
exactly how protein sequences evolve in the context of structure
and function. Thanks to increasing biodiversity in genomic studies,
progress is being made in detecting context-dependent variation
in substitution processes, but it remains unclear exactly what
features of the evolutionary process we should be looking for.
To address this, our laboratories have been developing a system
for simulating protein evolution in the context of structure
and function using lattice models of proteins and ligands (or
substrates). This system includes both thermodynamic features
of protein stability and population dynamics; we refer to this
approach as ab initio evolution to emphasize that the equilibrium
details of variant fitnesses arise from the physical principles
of the system, and not from any pre-conceived notions or arbitrary
mathematical distributions. Here, we discuss the relevance of
the system to evolutionary genomics and the choices that must
be made in trying to reproduce essential biological features
in the face of immense computational burdens. We present new
results on the coevolution during the divergence process and
retention of functionality in homologous recombinants following
population divergence. The designability, or sequence space
available to a structure, plays a key role in divergence and
recombinant function. These results have implications for understanding
viral evolution, speciation, and directed evolutionary experiments.
We also show that the results of our analysis of the divergence
process can guide improved methods for accurately approximating
folding probabilities in more complex systems that would otherwise
be beyond computational feasibility.
Sequences and protein
structures are congruent with functional and fitness differences
among Colias phosphoglucose isomerase genotypes
Wheat CW, Watt WB, Pollock DD, Schulte PM
The enzyme phosphoglucose isomerase, PGI, of Colias butterflies
(Lepidoptera, Pieridae) displays a widespread allozyme polymorphism.
Many studies on the biochemical function, organismal performance,
and fitness effects of Colias PGI genotypes have given evidence
of strong natural selection in the wild to maintain this polymorphism.
Here we begin to study the mechanism underlying this adaptive
polymorphism at the level of molecular sequence and structure.
The common electrophoretically-detectable alleles differ at
multiple amino acid positions, and also show some cryptic charge-neutral
amino acid variation hidden within the electrophoretic allele
classes. Structural modeling shows that all changes are at or
near PGI’s surface, and several naturally abundant variants
that distinguish these alleles are so placed as potentially
to alter subunit interaction and catalytic center geometry.
There is a large excess of intraspecific variation, both synonymous
and nonsynonymous, compared to interspecific fixation: there
are no fixed synonymous differences between species, and only
two fixed nonsynonymous differences. The fixed differences may
be due to positive selection, but sliding window analysis of
synonymous nucleotide diversity and Tajima’s D shows that
that the amino acid sites predicted to be foci of selection
based on structural and functional considerations also coincide
with the regions of highest synonymous diversity. They are thus
the most likely targets of balancing selection based on both
genetic and biochemical considerations. Colias' PGI gene, with
1668 bp of cDNA, is divided into 12 exons, spread over ~ 11kb
of chromosomal DNA, and intragenic recombination has been active
over much of the gene. Our results show that the relaxation
of constraint against amino acid variation, as one moves from
the interior cores of proteins to their surface, allows adaptive,
as well as neutral, natural variation to occur near or at those
surfaces. This case study of persistent polymorphism now offers
the integration of the genomic and molecular-structural bases
of natural variation with its consequences for metabolic and
organismal performance, thence for fitness, in wild populations.
37: NHGRI White Paper 2005
Proposal to sequence the first reptilian
genome: the Green Anole Lizard, Anolis carolinensis
J. Losos, E. Braun, D. Brown, S. Clifton, S. Edwards, J.
Gibson-Brown, T. Glenn, L. Guillette, D. Main, P. Minx, W. Modi,
M. Pfrender, D. Pollock, D. Ray, A. Shedlock, and W. Warren
Evolution
of base substitution gradients in primate mitochondrial genomes
Raina SZ, Faith JJ, Seligmann H, Disotell T, Stewart C-B,
and Pollock DD
Substitution patterns among nucleotides are often assumed to
be constant in phylogenetic analyses. Although variation in
the average rate of substitution among sites is commonly accounted
for, variation in the relative rates of specific types of substitution
are not. Here, we review details of methodologies used for detecting
and analyzing differences in substitution processes among predefined
groups of sites. We describe how such analyses can be performed
using existing phylogenetic tools, and discuss how new phylogenetic
analysis tools we have recently developed can be used to provide
more detailed and sensitive analyses, including study of the
evolution of mutation and substitution processes. As an example
we consider the mitochondrial genome, for which two types of
transition deaminations (C=>T and A=>G) are strongly
affected by single-strandedness during replication, resulting
in an asymmetric mutation process. Since time spent single-stranded
varies along the mitochondrial genome, their differential mutational
response results in very different substitution patterns in
different regions of the genome.
The beetle gut: a hyperdiverse
source of novel yeasts
Suh S-O, McHugh, JV, Pollock DD, Blackwell M
We isolated over 650 yeasts over a three year period from the
gut of a variety of beetles and characterized them on the basis
of LSU rDNA sequences and morphological and metabolic traits.
Of these, at least 200 were undescribed taxa, a number equivalent
to almost 30% of all currently recognized yeast species. A Bayesian
analysis of species discovery rates predicts further sampling
of previously sampled habitats could easily produce another
100 species. The sampled habitat is, thereby, estimated to contain
well over half as many more species as are currently known worldwide.
The beetle gut yeasts occur in 45 independent lineages scattered
across the yeast phylogenetic tree, often in clusters. The distribution
suggests that some of the yeasts diversified by a process of
horizontal transmission in the habitats and subsequent specialization
in association with insect hosts. Evidence of specialization
comes from consistent association over time and broad geographical
ranges of certain yeasts and beetle species. The discovery of
high yeast diversity in a previously unexplored habitat is a
first step toward investigating the basis of the interactions
and their impact in relation to ecology and evolution.
Modeling protein evolution has been frustratingly simplistic
in the past, but new methodologies and approaches have been
rapidly changing this situtation. Increased computational power,
improved phylogeny-based maximum likelihood and Bayesian statistics,
larger data sets, and better protein structure prediction methods
are jointly improving the outlook and allowing researchers to
improve the biological realism of protein models. They are also
allowing more detailed analysis of differences in processes
among sequence positions over space and time, of selection and
adaptation, coevolution, and functional divergence, and of ancestral
changes in function. The future is expected to bring improved
integration of models of protein evolution with protein structure
prediction, with the potential to dramatically improve the accuracy
and power of both
Context dependence and
coevolution among amino acid residues in proteins
Wang ZO and Pollock DD
As complete genomes accumulate, and the generation of genomic
biodiversity proceeds at an accelerating pace, the need to understand
the interaction between sequence evolution and protein structure
and function rises in prominence. The pattern and pace of substitutions
in proteins can provide important clues to functional importance,
functional divergence, and adaptive response. Coevolution between
amino acid residues and the context-dependence of the evolutionary
process are often ignored, however, due to their complexity;
but they are of critical importance for the accurate interpretation
of reconstructed evolutionary events. Since residues interact
with one another, and because the effect of substitutions can
depend on the structural and physiological environment in which
they occur, an accurate science of evolutionary functional genomics
and a complete understanding of selection in proteins requires
a better understanding of how context dependence affects protein
evolution. Here, we present new evidence from vertebrate cytochrome
oxidase sequences that pairwise coevolutionary interactions
between protein residues are highly dependent on tertiary and
secondary structure. We also discuss theoretical predictions
that impinge on our expectations of how protein residues may
interact over long distances due to their shared need to maintain
protein stability.
Analysis of among-site
variation in substitution patterns
Krishnan NM, Raina SZ, and Pollock DD
Substitution patterns among nucleotides are often assumed to
be constant in phylogenetic analyses. Although variation in
the average rate of substitution among sites is commonly accounted
for, variation in the relative rates of specific types of substitution
are not. Here, we review details of methodologies used for detecting
and analyzing differences in substitution processes among predefined
groups of sites. We describe how such analyses can be performed
using existing phylogenetic tools, and discuss how new phylogenetic
analysis tools we have recently developed can be used to provide
more detailed and sensitive analyses, including study of the
evolution of mutation and substitution processes. As an example
we consider the mitochondrial genome, for which two types of
transition deaminations (C=>T and A=>G) are strongly
affected by single-strandedness during replication, resulting
in an asymmetric mutation process. Since time spent single-stranded
varies along the mitochondrial genome, their differential mutational
response results in very different substitution patterns in
different regions of the genome.
Detecting gradients
of asymmetry in site-specific substitutions in mitochondrial
genomes
Krishnan NM, Seligmann H, Raina SZ, and Pollock DD
During mitochondrial replication, spontaneous mutations occur
and accumulate asymmetrically during the time spent single-stranded
by the heavy strand (DssH). The predominant mutations appear
to be deaminations from adenine to hypoxanthine (A=>H, which
leads to an A=>G substitution) and cytosine to thymine (C=>T).
Previous findings indicated that C=>T substitutions accumulate
rapidly and then saturate at high DssH, suggesting protection
or repair, whereas A=>G accumulates linearly with DssH. We
describe here the implementation of a simple hidden Markov model
(HMM) of among-site rate correlations to provide an almost continuous
profile of the asymmetry in substitution response for any particular
substitution type. We implement this model using a phylogeny-based
Bayesian Markov chain Monte Carlo (MCMC) approach. We compare
and contrast the relative asymmetries in all twelve possible
substitution types, and find that the observed transition substitution
responses determined using our new method agree quite well with
previous predictions of a saturating curve for C=>T transition
substitutions and a linear accumulation of A=>G transitions.
The patterns seen in transversion substitutions show much lower
among-site variation and are non-linear and more complex than
those seen in transitions. We also find that, after accounting
for the principal linear effect, some of the residual variation
in A=>G/G=>A response ratios is explained by the average
predicted nucleic acid secondary structure propensity at a site,
possibly due to protection from mutation when secondary structure
forms.
The ambush hypothesis:
Hidden stop codons prevent off-frame gene reading
Seligmann H and Pollock DD
Coding sequences lack stop codons, but many stops appear off-frame.
Off-frame stops (stops in -1 and +1 shifted reading frames,
termed hidden stops) terminate frameshifted translation, potentially
decreasing energy and resource waste on non-functional proteins.
Benefits may include reduced waste elimination costs and avoidance
of potentially cytotoxic frame-shifted products. Our “ambush”
hypothesis suggests that hidden stops are sometimes selected
for. Codons of many amino acids can contribute to hidden stops,
depending on the synonymous position state and adjacent codons.
In vertebrate mitochondria, 31.75% of all amino acid combinations
can form hidden stops. Codons with more potential to form hidden
stops have greater usage frequency and bias in their favor among
synonymous codons. Among primates, predicted mitochondrial rRNA
secondary structure stability correlates negatively with the
number of hidden stops in the mitochondrial genome. The taxonomic
distribution of genetic codes suggests that +1 frameshifts might
be more frequent than –1 frameshifts. This is confirmed
by analyses of primate mitochondrial genomes: species with unstable
rRNAs have more +1 stops, but the correlation is weak for -1
stops. High hidden stop density seems to be an adaptation in
species with slippage prone ribosomes (unstable rRNAs). Hidden
stops may thus compensate for reduced efficiency of some parts
of the biosynthetic machinery. Some experimental data confirm
our hypothesis: gene expression increases with the experimentally
manipulated number of stops in the promoter region of a gene,
suggesting biotechnological applications.
Ancestral sequence reconstruction
in primate mitochondrial DNA: compositional bias and effect
on functional inference
Krishnan NM, Seligmann H, Stewart, C-B, de Koning APJ, and Pollock
DD
Reconstruction of ancestral DNA and amino acid sequences is
an important means of inferring information about past evolutionary
events. Such reconstructions suggest changes in molecular function
and evolutionary processes over the course of evolution, and
are used to infer adaptation and convergence. Maximum likelihood
(ML) is generally thought to provide relatively accurate reconstructed
sequences compared to parsimony, but both methods lead to the
inference of multiple directional changes in nucleotide frequencies
in primate mitochondrial DNA (mtDNA). To better understand this
surprising result, as well as to better understand how parsimony
and ML differ, we constructed a series of computationally simple
“conditional pathway” methods that differed in the
number of substitutions allowed per site along each branch,
and also evaluated the entire Bayesian posterior frequency distribution
of reconstructed ancestral states. We analyzed primate mitochondrial
cytochrome b (Cyt-b) and cytochrome oxidase subunit I (COI)
genes and found that ML reconstructs ancestral frequencies that
are often more different from tip sequences than are parsimony
reconstructions. In contrast, frequency reconstructions based
on the posterior ensemble more closely resemble extant nucleotide
frequencies. Simulations indicate that these differences in
ancestral sequence inference are probably due to deterministic
bias caused by high uncertainty in the optimization-based ancestral
reconstruction methods (parsimony, ML, Bayesian maximum a posteriori).
In contrast, ancestral nucleotide frequencies based on an average
of the Bayesian set of credible ancestral sequences are much
less biased. The methods involving simpler conditional pathway
calculations have slightly reduced likelihood values compared
to full likelihood calculations, but can provide fairly unbiased
nucleotide reconstructions and may be useful in more complex
phylogenetic analyses than considered here due to their speed
and flexibility. To determine whether biased reconstructions
using optimization methods might affect inferences of functional
properties, ancestral primate mitochondrial tRNA sequences were
inferred and helix-forming propensities for conserved pairs
were evaluated in silico. For ambiguously reconstructed nucleotides
at sites with high base composition variability, ancestral tRNA
sequences from Bayesian analyses were more compatible with canonical
base pairing than were those inferred by other methods. Thus,
nucleotide bias in reconstructed sequences apparently can lead
to serious bias and inaccuracies in functional predictions.
Estimating the degree
of saturation in mutant screens
Pollock DD and Larkin J
Large-scale screens for loss-of-function mutants have played
a significant role in recent advances in developmental biology
and other fields. In such mutant screens, it is desirable to
estimate the degree of “saturation” of the screen
(i.e., what fraction of the possible target genes have been
identified). We applied Bayesian and maximum likelihood methods
for estimating the number of loci remaining undetected in large-scale
screens, and produce credibility intervals to assess the uncertainty
of these estimates. Since different loci may mutate to alleles
with detectable phenotypes at different rates, we also incorporated
variation in the degree of mutability among genes, using either
gamma-distributed mutation rates or multiple discrete mutation
rate classes. We examined eight published data sets from large-scale
mutant screens and find that credibility intervals are much
broader than implied by previous assumptions about the degree
of saturation of screens. The likelihood methods presented here
are a significantly better fit to data from published experiments
than estimates based on the Poisson distribution, which implicitly
assumes a single mutation rate for all loci. The results are
reasonably robust to different models of variation in the mutability
of genes. We tested our methods against mutant allele data from
a region of the Drosophila melanogaster genome for which there
is an independent genomics-based estimate of the number of undetected
loci, and found that the number of such loci falls within the
predicted credibility interval for our models. The methods we
have developed may also be useful for estimating the degree
of saturation in other types of genetic screens in addition
to classical screens for simple loss-of-function mutants, including
genetic modifier screens and screens for protein-protein interactions
using the yeast two-hybrid method.
27: Human Genomics 2004; 1(2): 85
Human genomics and the
role of evolutionary genomics
Pollock DD
Human Genomics has, from its outset, included a great deal of
evolutionary analysis. The structure of the editorial board
has representation from many evolution-based disciplines, including
population and quantitative genetics, and of course, evolutionary
genomics. This inclusion is the result of an obvious trend in
the field of genomics to incorporate more and more evolutionary
analysis, not just as an extra frill, but as a central component
of the field. The world now has over one hundred complete bacterial
genomes, and with human, roundworm, multiple fruitflies, mosquito,
rice, Arabidposis, pufferfish, mouse, rat, dog, chimpanzee,
chicken, and a growing number of other multicellular organisms
either sequenced or imminent, comparative genomics is coming
into its own. Still, one might argue that a journal of Human
Genomics should focus on its main target, Homo sapiens, and
leave aside mucking about with the multitude of other species
on the planet, most of which many self-respecting Homo sapiens
individuals might rather target with the bottom of their shoe
rather than with a multimillion dollar sequencing project. As
the evolutionary genomics editor, it seems necessary to provide
some explanation and justification.
Likelihood analysis of
asymmetrical mutation bias gradients in vertebrate mitochondrial
genomes
Faith JJ and Pollock DD
Protein-coding genes in mitochondrial genomes have varying degrees
of asymmetric skew in base frequencies at the third codon position.
The variation in skew among genes appears to be caused by varying
durations of time that the heavy strand spends in the mutagenic
single stranded state during replication (DssH). The primary data
used to study skew has been the gene-by-gene base frequencies
in individual taxa, which provides little information on exactly
what kinds of mutations are responsible for the base frequency
skew. To assess the contribution of individual mutation components
to the ancestral vertebrate substitution pattern, here we analyze
a large data set of complete vertebrate mitochondrial genomes
in a phylogeny-based likelihood context. This also allows us to
evaluate the change in skew continuously along the mitochondrial
genome, and to directly estimate relative substitution rates.
Our results indicate that different types of mutation respond
differently to the gradient. A primary role for hydrolytic deamination
of cytosines in creating variance in skew among genes was not
supported, but rather linearly increasing rates of mutation from
adenine to hypoxanthine with appear to drive regional differences
in skew. Substitutions due to hydrolytic deamination of cytosines,
although common, appear to quickly saturate, possibly due to stabilization
by the mitochondrial DNA single strand binding protein. These
results should form the basis of more realistic models of DNA
and protein evolution in mitochondria.
25: NHGRI White Paper 2003
Proposal for complete sequencing of the genome
of a Marsupial, the gray, short-tailed opossum, Monodelphis
domestica
Amemiya CT, Greally JM, Jirtle RL, Lander ES, Lindblad-Toh
K, Miller RD, Pollock DD, Samallow PB, Springer MS, and Wilson RK
Metatherian (“marsupial”) mammals are phylogenetically
distinct from current mammalian biomedical models, all of which
are eutherian (“placental”) species. However, marsupials
and eutherians are more closely related to one another than to
any other vertebrate model species (i.e., birds, amphibians, fishes).
Fossil evidence establishes a minimum date of 125 million years
(MY) for the separation of eutherian and metatherian mammals (JI
et al. 2002), while analyses of nuclear gene sequences suggest
that metatherian / eutherian divergence may be as old as 173-190
MY (KUMAR and HEDGES 1998; WOODBURNE et al. 2003). To place this
in context, the evolutionary gulf between mammals and the next
most closely related group of non-mammalian research models, i.e.,
birds (chicken), is approximately 300 –350 MY. Thus, the
marsupial – eutherian relationship represents a unique midpoint
in age relative to existing mammalian and non-mammalian vertebrate
models. As a legacy of their common ancestry, marsupials and eutherians
share basic genetic mechanisms and molecular processes that represent
fundamental (ancient) mammalian characteristics. Nevertheless,
since their divergence, eutherian and marsupial mammals have evolved
many distinctive morphologic, physiologic, and genetic variations
on these elemental mammalian designs. These phylogenetically restricted
differences can be used as comparative tools for examining the
underlying molecular and genetic processes that are common to
all mammalian species, and thereby help to reveal how variations
in these mechanisms lead to differences in gene regulation, expression,
and function. As the closest sister group to eutherian mammals,
marsupials are also the most appropriate “outgroup”
for assessing the relative antiquity or novelty of the molecular
and genetic changes that have occurred among the many eutherian
species (including ourselves) presently used in biomedical and
evolutionary research..
24: Journal of Molecular Evolution 2003; 56(4): 375-376
The Zuckerkandl Prize:
Structure and Evolution
Pollock DD
Guest Editorial: The Zuckerkandl Prize, established by Springer-Verlag
in 2002 to honor Emile Zuckerkandl and his contributions to molecular
evolution, goes this year to Gustavo Caetano-Anollés for
his paper on “Evolved RNA Secondary Structure and the rooting
of the Universal Tree of Life” (Caetano-Anollés 2002).
The editors of the Journal of Molecular Evolution have judged
this to be the best paper in the journal last year due to its
creative use of structure, and the evolution of structure, to
reconstruct deep phylogenies.
Is sparse taxon sampling
a problem for phylogenetic inference?
Hillis, DM, Pollock DD, McGuire JA, and Zwickl DJ
No abstract: ...There is no simple answer to the question posed
in the heading of this section; the answer will depend on the
particular situation being examined (the scope of the problem,
the number of taxa already sequenced, the number of characters
already collected, and the quantity and the availability of
additional relevant taxa to include). We disagree with the assertion
of Rosenberg and Kumar (2002) that more characters per taxon
is necessarily a better strategy than more taxa for the same
characters. Rosenberg and Kumar (2002) put ther argument in
terms of the current genome sequencing studies, in which many
genes (or complete genomes) are examined from very few taxa.
Rosenberg and Kumar 92002) argued that their conclusions "mesh
well" with this scattered genome approach. In contrast,
we propose that this approach will likely result in poorly estimated
evolutionary models, poorly estimated evolutionary trees, and
a poor overall view of evolutionary history. If one is interested
in inferring the evolutionary history of life, a much broader
sample of taxa (perhaps sequence for far less than full genomes)
will result in a much more accurate estimate of phylogeny than
will complete genomes of only a small sample of taxa.
Increased taxon sampling
is advantageous for phylogenetic inference
Pollock DD, Zwickl DJ, McGuire JA, and Hillis DM
Until recently, it was believed that complex phylogenies might
be extremely difficult to reconstruct due to the phenomenal rate
of increase in the number of possible phylogenies as the number
of taxa increases. However, Hillis (1996) showed through simulation
that, for at least one complex phylogeny of angiosperms with 228
taxa, reconstruction was far more accurate than expected, even
with relatively modest amounts of DNA sequence data. This led
to a flurry of papers on the subject of taxon sampling and phylogenetic
reconstruction, with focus quickly shifting from the question
of whether complex phylogenies can be reconstructed to whether
and how much an existing phylogeny can be improved through increased
taxon sampling (Hillis, 1998; Kim, 1998; Poe, 1998; Poe and Swofford,
1999; Pollock and Bruno, 2000; Rannala et al., 1998; Yang, 1998).
Although a statistician might intuitively believe that it is generally
better (or at least no worse) to increase the amount of data to
resolve a question in statistical inference, the benefits of taxon
addition for phylogenetic inference remain controversial. ...A
recent paper on the subject of taxon addition (Rosenberg and Kumar,
2001) concludes that increased taxon sampling is of little benefit
to phylogenetic inference when compared to increasing sequence
length. We disagree with their interpretation and believe that
their data support the importance of increased taxon sampling.
In addition, some of their data were simulated under extreme conditions
(i.e., substitution rates that were very high or low, or sequences
that were unreasonably short). Large error values and non-linear
relationships at these extremes make it difficult to interpret
effects for the majority of the range, and averaging across the
entire range is inappropriate. Moreover, we do not believe that
Rosenberg and Kumar (2001) used the most appropriate metric to
measure the relative effect of taxon addition. Our reanalysis
of their simulated data indicates that increased taxon sampling
is highly beneficial for phylogenetic inference..
Genomic biodiversity,
phylogenetics, and coevolution in proteins
Pollock DD.
Comprehensive sampling of genomic biodiversity is fast becoming
a reality for some genomic regions and complete organelle genomes.
Genomic biodiversity is defined as large genomic sequences from
many species, and here some recent work is reviewed that demonstrates
the potential benefits of genomic biodiversity for molecular evolutionary
analysis and phylogenetic reconstruction. This work shows that,
using likelihood-based approaches, taxon addition can dramatically
improve phylogenetic reconstruction. Features, or dynamics, of
the evolutionary process are much more easily inferred with large
numbers of taxa, and large numbers are essential for discriminating
differences in evolutionary patterns between sites. Accurate prediction
of site-specific patterns can improve phylogenetic reconstruction
by an amount equivalent to quadrupling sequence length. Genomic
biodiversity is particularly central to research relating patterns
of evolution, adaptation, and coevolution to structural and functional
features of proteins. Research on detecting coevolution between
amino acid residues in proteins is reviewed that demonstrates
a clear need for much greater numbers of closely related taxa
to better discriminate site-specific patterns of interaction,
and to allow more detailed analysis of coevolutionary interactions
between subunits in protein complexes. It is argued that parsing
out coevolutionary and other context-dependent substitution probabilities
is essential for discriminating between coevolution and adaptation,
and for more realistically modeling the evolution of proteins.
Research is also reviewed that argues for increasing the efficiency
of acquiring genomic biodiversity, and suggests that this might
be done by simultaneously shotgun cloning and sequencing genomic
mixtures from many species. Increased efficiency is a prerequisite
if genomic biodiversity levels are to rapidly increase by orders
of magnitude, and thus lead to dramatically improved understanding
of interactions between protein structure, function, and sequence
evolution.
All of biology is based on evolution. Evolution is the organizing
principle for understanding the shared history of all biological
organisms. Evolution describes the similarities between different
organisms, as well as explaining how differences emerged. In addition
to answering basic questions about the history of life, evolutionary
perspectives and information drawn from evolutionary analyses
can provide information highly relevent to many biological, biotechnological,
and biomedical problems. There is also growing interest in mimicking
evolution in the test tube in order to develop RNA, proteins,
and organisms with specified properties.
We study the evolution of protein functionality using a two-dimensional
lattice model. The characteristics particular to evolution, such
as population dynamics and early evolutionary trajectories, have
a large effect on the distribution of observed structures. Only
subtle differences are observed between the distribution of structures
evolved for function and those evolved for their ability to form
compact structures.
Structures, phylogenies, and genomes: The integrated
study of protein evolution
Goldstein RA, Pollock DD, and Thorne JL
For the past decades, evolutionary biologists have tried to reconstruct
evolutionary histories, to piece together phylogenetic trees,
and to understand the network of hereditary relationships. Such
approaches (whether it is admitted or not) are based on models
of the evolutionary process. These tasks would be easier if reality
would better match the simplest models. Unfortunately for these
scientists, evolution takes place in a complicated web of constraints,
with changes in the DNA sometimes but not always translating to
changes in amino acids which may or may not result in significant
changes in the properties of these expressed proteins. All of
this occurs in a complicated and interconnected fitness landscape,
where different locations in the protein may be under radically
different selective pressure. This situation has led a number
of investigators to bring more of the biologial and biochemical
complexity into these evolutionary models, to develop approaches
with a closer fidelity to biological reality with the hope that
more accurate pictures of biological history will result.
Assessing an unknown
evolutionary process: effect of increasing site-specific knowledge
through taxon addition
Pollock DD, Bruno WJ.
Assessment of the evolutionary process is crucial for understanding
the effect of protein structure and function on sequence evolution
and for many other analyses in molecular evolution. Here, we used
simulations to study how taxon sampling affects accuracy of parameter
estimation and topological inference in the absence of branch
length asymmetry. With maximum-likelihood analysis, we find that
adding taxa dramatically improves both support for the evolutionary
model and accurate assessment of its parameters when compared
with increasing the sequence length. Using a method we call "doppelganger
trees," we distinguish the contributions of two sources of
improved topological inference: greater knowledge about internal
nodes and greater knowledge of site-specific rate parameters.
Surprisingly, highly significant support for the correct general
model does not lead directly to improved topological inference.
Instead, substantial improvement occurs only with accurate assessment
of the evolutionary process at individual sites. Although these
results are based on a simplified model of the evolutionary process,
they indicate that in general, assuming processes are not independent
and identically distributed among sites, more extensive sampling
of taxonomic biodiversity will greatly improve analytical results
in many current sequence data sets with moderate sequence lengths.
A case for evolutionary
genomics and the comprehensive examination of sequence biodiversity
Pollock DD, Eisen JA, Doggett NA, Cummings MP.
Comparative analysis is one of the most powerful methods available
for understanding the diverse and complex systems found in biology,
but it is often limited by a lack of comprehensive taxonomic sampling.
Despite the recent development of powerful genome technologies
capable of producing sequence data in large quantities (witness
the recently completed first draft of the human genome), there
has been relatively little change in how evolutionary studies
are conducted. The application of genomic methods to evolutionary
biology is a challenge, in part because gene segments from different
organisms are manipulated separately, requiring individual purification,
cloning, and sequencing. We suggest that a feasible approach to
collecting genome-scale data sets for evolutionary biology (i.e.,
evolutionary genomics) may consist of combination of DNA samples
prior to cloning and sequencing, followed by computational reconstruction
of the original sequences. This approach will allow the full benefit
of automated protocols developed by genome projects to be realized;
taxon sampling levels can easily increase to thousands for targeted
genomes and genomic regions. Sequence diversity at this level
will dramatically improve the quality and accuracy of phylogenetic
inference, as well as the accuracy and resolution of comparative
evolutionary studies. In particular, it will be possible to make
accurate estimates of normal evolution in the context of constant
structural and functional constraints (i.e., site-specific substitution
probabilities), along with accurate estimates of changes in evolutionary
patterns, including pairwise coevolution between sites, adaptive
bursts, and changes in selective constraints. These estimates
can then be used to understand and predict the effects of protein
structure and function on sequence evolution and to predict unknown
details of protein structure, function, and functional divergence.
In order to demonstrate the practicality of these ideas and the
potential benefit for functional genomic analysis, we describe
a pilot project we are conducting to simultaneously sequence large
numbers of vertebrate mitochondrial genomes.
The genomic data available to computational biologists represents
the product of the complex processes of evolution. In particular,
the forces of mutation, duplication, and selection have acted
to sculpt modern protein sequence and structure in the context
of changing functional requirements. Just as crystallographers
are able to determine protein structures through an analysis of
X-ray diffraction patterns, scientists are learning to read the
evolutionary history of proteins in order to infer and explain
both structure and function. This pursuit depends on the development
of new computational approaches in order to make optimal use of
genomic data, and requires interaction with experiment for comparison
and verification of computational results.
Coevolving protein
residues: maximum likelihood identification and relationship to
structure
Pollock DD, Taylor WR, and Goldman N
The identification of protein sites undergoing correlated evolution
(coevolution) is of great interest due to the possibility that
these pairs will tend to be adjacent in the three-dimensional
structure. Identification of such pairs should provide useful
information for understanding the evolutionary process, predicting
the effects of site-directed substitution, and potentially for
predicting protein structure. Here, we develop and apply a maximum
likelihood method with the aim of improving detection of coevolution.
Unlike previous methods which have had limited success, this method
allows for correlations induced by phylogenetic relationships
and for variation in rate of evolution along branches, and does
not rely on accurate reconstruction of ancestral nodes. In order
to reduce the complexity of coevolutionary relationships and identify
the primary component of pairwise coevolution between two sites,
we reduce the data to a two-state system at each site, regardless
of the actual number of residues observed at that site. Simulations
show that this strategy is good at identifying simple correlations
and at recognizing cases in which the data are insufficient to
distinguish between coevolution and spurious correlations. The
new method was tested by using size and charge characteristics
to group the residues at each site, and then evaluating coevolution
in myoglobin sequences. Grouping based on physicochemical characteristics
allows categorization of coevolving sites into positive and negative
coevolution, depending on the correlation between equilibrium
state frequencies. We detected a striking excess of negative coevolution
(corresponding to charge) at sites brought into proximity by the
periodicity of the alpha-helix, and there was also a tendency
for sites with significant likelihood ratios to be close in the
three-dimensional structure. Sites on the surface of the protein
appear to coevolve both when they are close in the structure,
and when they are distant, implying a role for folding and/or
avoidance of quaternary structure in the coevolution process.
Copyright 1998 Academic Press.
Increased accuracy in
analytical molecular distance estimation
Pollock DD
Analytical molecular distance estimates can be inaccurate and
biased estimates of the total number of substitutions not only
when the model of evolution they are based on is incorrect, but
also when the method of estimating the total is too simple. This
comes about because when there are different types of substitutions
occurring simultaneously, it can become extremely difficult to
estimate the number of the more quickly evolving type, and the
variance of this larger number can overwhelm the total estimate.
In this paper, in an extension of earlier work with a simple two-parameter
model of evolution, more accurate analytical distances are derived
for models appropriate to a variety of known DNA types using generalized
least squares principles of noise reduction. It is shown that
the new estimates can be applied to achieve more accurate results
for site-to-site rate variation, regions with biased nucleotide
frequencies, and synonymous sites in protein-coding regions. This
study also includes a methodology to obtain accurate distance
estimates for large numbers of sequence regions evolving in different
manners. Copyright 1998 Academic Press.
Microsatellite behavior with range
constraints: parameter estimation and improved distances for use
in phylogenetic reconstruction
Pollock DD, Bergman A, Feldman MW, Goldstein DB
A symmetric stepwise mutation model with reflecting boundaries
is employed to evaluate microsatellite evolution under range constraints.
Methods of estimating range constraints and mutation rates under
the assumptions of the model are developed. Least squares procedures
are employed to improve molecular distance estimation for use
in phylogenetic reconstruction in the case where range constraints
and mutation rates vary across loci. The bias and accuracy of
these methods are evaluated using computer simulations, and they
are compared to previously existing methods which do not assume
range constraints. Range constraints are seen to have a substantial
impact on phylogenetic conclusions based on molecular distances,
particularly for more divergent taxa. Results indicate that if
range constraints are in effect, the methods developed here should
be used in both the preliminary planning and final analysis of
phylogenetic studies employing microsatellites. It is also seen
that in order to make accurate phylogenetic inferences under range
constraints, a larger number of loci are required than in their
absence.
Molecular phylogeny for Colias
butterflies and their relatives (Lepidoptera: Pieridae)
Pollock DD, Watt WB, Rashbrook VK, Iyengar EV
The sulfur butterflies, Colias spp., and their relatives in the
family Pieridae have been the subjects of diverse behavioral,
ecological, and evolutionary studies. However, their phylogeny
is uncertain in many respects. We used DNA sequences from 2 mitochondrial
gene blocks, 333 bp of the cytochrome oxidase I subunit (CO I)
and 1,261 bp from the 2 ribosomal genes and the tRNA between them
(rDNA), as character sources to test existing phylogenetic hypotheses
and begin to infer others. The rDNA block resolves better at deeper
nodes of the phylogeny, and the CO I block at shallower nodes.
Our results support sister status for subfamilies Coliadinae and
Pierinae within Pieridae; independent tribal status for Euchloini
and Pierini within Pierinae; status as sister genera for Colias
and Zerene within Coliadinae; and monophyly within subgenus C.
(Euoolias) of all North American Colias studied. Our results suggest
that the Neotropical coliad genus Eurema may warrant splitting,
as some early workers proposed, but do not support the recently
proposed splitting of Eurasian C. erate from subgenus C. (Eriocolias)
into the separate subgenus C. (Neocolias).
Effectiveness of correlation
analysis in identifying protein residues undergoing correlated
evolution
Pollock DD, Taylor WR.
Various methods for detecting correlation between sites were evaluated
by ascertaining their ability to discriminate positively correlated
sites from background correlation at randomly evolved sites. A
model for generating pairwise correlations of different degrees
is also described. An assortment of physicochemical vectors and
similarity and difference matrices were used to discriminate correlated
change. There was little difference in effectiveness between the
different matrices, but there were significant differences between
the matrices and the physicochemical vectors. It is shown that
all methods investigated exhibit significant inability to screen
out background correlation, particularly in the presence of phylogenetic
relatedness between the sequences. Methods using the matrices
are unable to distinguish positively correlated from negatively
correlated, or compensatory, replacements.
Microsatellite genetic distances
with range constraints: analytic description
and problems of estimation
Feldman MW, Bergman A, Pollock DD, Goldstein DB.
Statistical properties of the symmetric stepwise-mutation model
for microsatellite evolution are studied under the assumption
that the number of repeats is strictly bounded above and below.
An exact analytic expression is found for the expected products
of the frequencies of alleles separated by k repeats. This permits
characterization of the asymptotic behavior of our distances D1
and (delta mu)2 under range constraints. Based on this characterization
we develop transformations that partially restore linearity when
allele size is restricted. We show that the appropriate transformation
cannot be applied in the case of varying mutation rates (beta)
and range constraints (R) because of statistical difficulties.
In the special case of no variation in beta and R across loci,
however, the transformation simplifies to a usable form and results
in a distance much more linear with time than distances developed
for an infinite range. Although analytically incorrect in the
case of variation in beta and R, the simpler transformation is
surprisingly insensitive to variation in these parameters, suggesting
that it may have considerable utility in phylogenetic studies.
A comparison of two
methods for constructing evolutionary distances from a weighted
contribution of transition and transversion differences
Pollock DD, Goldstein DB.
Since the initial work of Jukes and Cantor (1969), a number of
procedures have been developed to estimate the expected number
of nucleotide substitutions corresponding to a given observed
level of nucleotide differentiation assuming particular evolutionary
models. Unlike the proportion of different sites, the expected
number of substitutions that would have occurred grows linearly
with time and therefore has had great appeal as an evolutionary
distance. Recently, however, a number of authors have tried to
develop improved statistical approaches for generating and evaluating
evolutionary distances (Schoniger and von Haeseler 1993; Goldstein
and Polock 1994; Tajima and Takezaki 1994). These studies clearly
show that the estimated number of nucleotide substitutions is
generally not the best estimator for use in reconstruction of
phylogenetic relationships. The reason for this is that there
is often a large error associated with the estimation of this
number. Therefore, even though its expectation is correct (i.e.,
on average the expected number of substitutions is proportional
to time--but see Tajima 1993), it is not expected to be as useful
as estimators designed to have a lower variance.
Evolutionary relations among vertebrate muscle-type
lactate dehydrogenases
Quattro JM, Pollock DD, Powell M, Woods HA, Powers DA.
Gene duplication has produced two lactate dehydrogenase (LDH)
isozymes, LDH-A and LDH-B, that are found in essentially all vertebrates.
On the basis of the biochemical properties of the LDH-A and LDH-B
isozymes, it has been suggested that each locus is orthologous
among all vertebrates. However, phylogenetic studies have not
supported a common evolutionary history among the LDH-A isozymes,
particularly when those from lower vertebrates are examined. We
present here the sequence of a muscle-type LDH from Fundulus heteroclitus,
a teleost fish for which the LDH-B sequence has been determined
and shown to be unrelated phylogenetically to tetrapod LDH-A isozymes.
Although the sequence of the teleost muscle LDH shares certain
features with the LDH-A of tetrapods, phylogenetic analyses do
not support an orthologous relation among the LDH-A isozymes of
teleost fish and tetrapod vertebrates.
Least squares estimation
of molecular distance--noise abatement in phylogenetic reconstruction
Goldstein DB, Pollock DD.
Zuckerkandl and Pauling (1962, "Horizons in Biochemistry,"
pp. 189-225, Academic Press, New York) first noticed that the
degree of sequence similarity between the proteins of different
species could be used to estimate their phylogenetic relationship.
Since then models have been developed to improve the accuracy
of phylogenetic inferences based on amino acid or DNA sequences.
Most of these models were designed to yield distance measures
that are linear with time, on average. The reliability of phylogenetic
reconstruction, however, depends on the variance of the distance
measure in addition to its expectation. In this paper we show
how the method of generalized least squares can be used to combine
data types, each most informative at different points in time,
into a single distance measure. This measure reconstructs phylogenies
more accurately than existing non-likelihood distance measures.
We illustrate the approach for a two-rate mutation model and demonstrate
that its application provides more accurate phylogenetic reconstruction
than do currently available analytical distance measures.
3: Cytog. Cell. Genet 1991; 58(1-4): 1930
Chromosomal localization of the calbindin gene
Modi, W. S., M. Dean, D. D. Pollock, H. N. Suanez, and S. Christakos.
2: Cytog. Cell. Genet 1991; 58(1-4): 1870
Regional localization of the human glutaminase
gls and interleukin-9 il9 genes by in situ hybridization
Modi WS, Pollock DD, Mock BA, Banner C, Renauld JC, Van Snick
J.
Regional localization of the human glutaminase
GLS and interferon-9 IL9 genes by in-situ
hybridization
Modi WS, Pollock DD, Mock BA, Banner C, Renauld JC, and Van Snick
J
Phosphate-activated glutaminase is found in the mammalian small
intestine, brain, and kidney, but not in liver. The enzyme initiates
the catabolism of glutamine as the principal respiratory fuel
in the small intestine, may synthesize the neurotransmitter glutamate
in the brain, and functions in the kidney to help maintain systemic
pH homeostasis. Interleukin-9 (IL9) is a relatively new cytokine
that supports the growth of the helper T-cell clones, mast cells,
and megakaryoblastic leukemia cells. cDNA clones have recently
been obtained for each of these genes. The human loci for phosphate-activated
glutaminase (GLS) and IL9 have previously been mapped to chromosomes
2 and 5, respectively, by analysis of somatic cell hybrid DNAs.
By using chromosomal in situ hybridization, we have regionally
mapped GLS to 2q32 .fwdarw. q34 and IL9 to 5q31 .fwdarw. q35.